 These are rare neuroendocrine tumors that produce excess catecholamines (adrenaline and noradrenaline).
These are rare neuroendocrine tumors that produce excess catecholamines (adrenaline and noradrenaline).
While closely related, they differ in location.
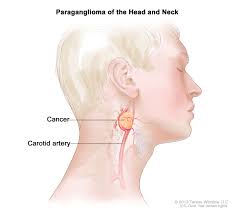
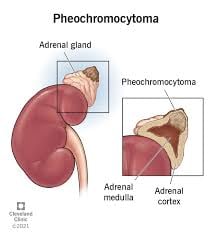
Pheochromocytoma (PHEO) and paraganglioma (PGL) are rare neuroendocrine tumors originating from chromaffin cells in the adrenal medulla (PHEO, ~80-85% of cases) or extra-adrenal sympathetic/parasympathetic sites (PGL).
P and P are diagnosed at about 2 to 3000 new cases per year in the US.
They overproduce catecholamines (e.g., norepinephrine, epinephrine), leading to endocrine and cardiovascular effects.
There are no histological, molecular, biochemical, or clinical aspects that can differentiate between benign and malignant tumors.
Most patients with P and P (75%) are not metastatic and therefore curable by surgery.
Most patients (~50-60%) are symptomatic due to catecholamine excess, forming the classic triad: Episodic or sustained hypertension (most common, >90%). Headache (severe, throbbing). Profuse sweating (diaphoresis).
Other signs include palpitations, anxiety, tremors, abdominal pain, or constipation. ~25% are asymptomatic, discovered incidentally on imaging.
In children/adolescents (5-10% of cases), symptoms may include fever or weight loss.
Familial cases (e.g., MEN2, VHL) often present earlier and bilaterally.
Incidence is 0.2-0.8 cases per 100,000 annually, with up to 40% hereditary (mutations in genes like SDHx, VHL, RET, NF1).
These tumors can be benign (90%) or malignant (10-15%), often presenting in ages 30-50.
Up to 25% are associated with metastasis and in 50% of those cases, the metastasis are present at the time of discovery of the primary tumor.
In the other 50%, the metastasis happen years with decades after discovery of the primary tumor.
The larger these tumors are, the higher the risk of metastases.
Sympathetic paragangliomas are mainly located in these chest, abdomen and pelvis, and the rate of metastasis is higher when compared with tumors that are coming from the head and neck areas.
Tumors associated with germline pathologic variants of the STHB gene are associated with metastasis more often when compared with all the molecular profiles in P and P’s.
P and P tumors are able to produce catecholamine hormones such as adrenaline and/or adrenaline that may predisposed the cardiovascular gastrointestinal disease leading to significant morbidity and mortality.
Pheochromocytoma: Catecholamine-producing tumor arising from chromaffin cells in the adrenal medulla Accounts for about 80-85% of these tumors
Paraganglioma:
Catecholamine-producing tumor arising from extra-adrenal chromaffin tissue-paraganglia. Can occur anywhere along the sympathetic or parasympathetic chain Common locations: abdomen, near aorta, chest, head and neck, bladder
Incidence: 2-8 per million per year
Can occur at any age, peak in 30s-50s
About 10-15% are malignant
Significant hereditary component: up to 30-40% have germline mutations
Clinical Presentation
Consider in any patient with resistant hypertension, or adrenal incidentaloma
The great mimic as it can present with diverse symptoms
A classic triad of occurs in <50% of patients.
Episodic headaches Profuse sweating Palpitations
Other Common Symptoms:
Hypertension is sustained in 50-60%, paroxysmal in 30%.
Pallor Tremor Anxiety or sense of impending doom Chest or abdominal pain Nausea and vomiting Weight loss Hyperglycemia
Paroxysmal episodes of catecholamine release causing:
Sudden severe hypertension Tachycardia Severe headache Profuse sweating Triggered by physical activity, abdominal pressure, certain foods like tyramine, medications, or occurring spontaneously
Some tumors are discovered incidentally on imaging and may be biochemically active without symptoms
Previously taught that 10% were:
– Bilateral – Extra-adrenal – Malignant – Familial – Pediatric
Modern understanding:-Hereditary cases are much more common (~40%), and malignancy rates vary by tumor type and genetics.
Approximately 25% of patients with pheochromocytoma or paraganglioma will eventually have metastatic disease.
Metastatic pheochromocytoma or paraganglioma is associated with lower survival than non-metastatic disease and has a high degree of complications owing to tumor burden, disease progression, and excessive catecholamine secretion leading to cardiovascular and gastrointestinal illness.
The five year survival among patients with metastatic pheochromocytoma or paraganglioma is approximately 60%.
Diagnosis
Diagnosis follows a stepwise approach per international guidelines:
Biochemical Testing: First-line plasma free metanephrines (sensitivity >96%) or 24-hour urine fractionated metanephrines/catecholamines. Avoid false positives from caffeine/stress.
Metanephrines and normetanephrines Catecholamines (epinephrine, norepinephrine) Vanillylmandelic acid (VMA) – less commonly used now
Fractionated metanephrines have >95% sensitivity
Imaging: CT/MRI for localization (sensitivity 90-100%). Functional imaging: 123I-MIBG scintigraphy or 68Ga-DOTATATE PET/CT for staging/metastases (preferred for PGL).
Genetic Testing: Recommended for all patients (NCCN/Endocrine Society guidelines), especially if young, bilateral, or family history.
Differential: Essential hypertension, anxiety disorders, or other tumors (e.g., neuroblastoma).
Plasma or 24-hour urine measurements:
Imaging after biochemical confirmation:
CT or MRI of abdomen/pelvis- first-line to locate tumor
Functional imaging: ¹²³I-MIBG scintigraphy (metaiodobenzylguanidine) ⁶⁸Ga-DOTATATE PET/CT (increasingly preferred) ¹⁸F-FDG PET/CT (for metastatic disease)
Genetic Testing is recommended for all patients due to high hereditary rate.
Associated Genetic Syndromes:
Multiple Endocrine Neoplasia Type 2 (MEN2)-RET mutations Von Hippel-Lindau (VHL) disease-this regulation of the hypoxia-inducible factor to alpha pathway is one of the key oncogenic drivers of metastatic, pheochromocytoma and paraganglioma. Neurofibromatosis Type 1 (NF1) Familial paraganglioma syndromes-SDHx mutations (SDHB, SDHD, SDHC, SDHA, SDHAF2) MAX, TMEM127, and others Most cases of metastatic, pheochromocytoma and paraganglioma are characterized by pseudohypoxia and up to 50% or associated with a STHB germline pathogenic variant.
Medical Management-Preoperative α-blockade
Currently, there is no standard treatment for unresectable, metastatic, pheochromocytoma, or paraganglioma.
Alpha-blockade
Phenoxybenzamine (non-selective, irreversible) Doxazosin or prazosin (selective α1-blockers) Started 7-14 days before surgery Liberal salt and fluid intake encouraged
Beta-blockade-if needed, only after adequate alpha-blockade.
Never give beta-blockers first because of risk of unopposed α-stimulation and hypertensive crisis.
For persistent tachycardia or arrhythmias
Other medications:
Calcium channel blockers Metyrosine (inhibits catecholamine synthesis) – in select cases
Alternative treatments include peptide receptor radionucleotide therapy (Lutetium LU 177 dotate, somatostatin, receptor analogues and chemotherapy with cyclophosphamide, vincristine, dacarbazine and temozolomide, and tying kinase inhibitors.
Belzutifan a hypoxia inducible factor inhibitor is an efficacious treatment for pheochromocytoma and paraganglioma.
Belzutifan shows 25-40% response rates in metastatic PPGL trials.
Surgical Treatment:
Primary treatment for localized disease
Laparoscopic adrenalectomy preferred for pheochromocytomas Open surgery for large tumors, paragangliomas, or suspected malignancy
Complete resection is curative for benign tumors
External beam radiation for bone metastases.
Perioperative considerations:
Experienced anesthesia team essential
Anticipate intraoperative hemodynamic instability
Postoperative hypotension common as may need fluids, pressor support
Common sites for metastases:lymph nodes, bone, liver, lung
Prognosis
Benign tumors:Excellent prognosis with complete surgical resection
Malignant tumors: 5-year survival ~40-50% Varies significantly by genetic mutation (SDHB mutations have worse prognosis) Can have indolent course with prolonged survival even with metastases
Follow-up Lifelong surveillance required:
Annual biochemical testing (plasma/urine metanephrines)
Periodic imaging if biochemically active
Screen for recurrence, metachronous tumors, or metastases
Family screening and genetic counseling for hereditary cases
High hereditary component – genetic testing for all
Biochemical cure confirmed by normalized metanephrines 2-6 weeks post-surgery
SDHB mutations associated with higher malignancy risk
Can occur during pregnancy with high maternal and fetal risk.
Tailored surveillance (e.g., annual imaging for SDHx carriers)
Prophylactic surgery in high-risk syndromes (e.g., RET mutation in MEN2).
Perioperative mortality <1% with proper blockade.
Long-term: 5-year survival >95% for benign, 40-77% for malignant.
Belzutifan associated with a 26% response rate with no complete responses and a clinical benefit in 85% of patients.