 Among the most malignant mesenchymal tumors of soft tissues.
Among the most malignant mesenchymal tumors of soft tissues.
Spindle cell tumors that arise from Schwann cells.
Includes schwannomas, neurogenic sarcomas, neurofibrosarcomas, anaplastic neurofibroma, and malignant neurilemoma.
See neurofibromatosis-1 (NF1).
Most patients have a truncating lesion in the NF1 gene on chromsome 17.
Accounts for 3-10% of soft tissue sarcomas, with 15-70% occurring in NF-1 patients.
Neurofibromatosis is the main risk factor for the development of this lesion.
Incidence in NF1 patient’s 2-29%
Incidence in the general population about 0.001%.
Develops in 4% of patients with neurofibromatosis type I.
Arises from pre-existing neurofibromas and subcutaneous/spinal nerve root neurofibromas but not from cutaneous neurofibromas in NF1 patients.
The risk of developing MPNSTs is increased 3 times in the presence of subcutaneous neurofibromas and 20 folds in the presence of internal neurofibromatosis.
10-50%MPNSTs result from malignant transformation of plexiform neurofibromas.
Cumulative risk of development of this type lesion in NF1 patient’s during your lifetime is 8-13%.
These lesions on the main cause of death in NF1 patients.
De novo lesions are the second most common types of lesions.
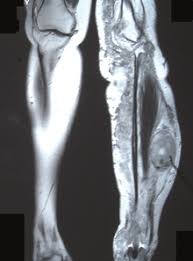
Can arise in any part of the body, but most commonly in the proximal upper and lower extremities and trunk.
Most patients are asymptomatic, but may have symptomatic mass with sensory or motor function loss, pain, visceral symptoms, and autonomic dysfunction.
The finding of a mass perpendicular to the axis of a peripheral nerve in combination with a positive Tinel’s sign suggests the presence of pa tumor of peripheral nerve origin.
Can rarely arise from malignant degeneration within a schwannoma, ganglioneuroma, or a pheochromocytoma.
Lesions arise typically in preexisting neurofibromas and are aggressive with frequent local recurrence and distant metastases.
Histologically the lesions have dense fascicles of spindle cells.
Median time from onset of symptoms to diagnosis about 5.5 months (Baehring).
Progressive pain, sensory or motor deficits and rapid growth in the size of the mass suggest the presence of a malignant lesion.
The presence of such a lesion requires ruling out the presence of NF1 or history of prior radiation exposure.
Lesions that are difficult to palpate suggest malignancy and tissue infiltration.
The size of the lesions does not indicate malignancy and can vary widely.
Radiation a common risk factor with almost 10% of lesions occurring in individuals following radiation, with an average lag time of 15 years.
MRI best noninvasive diagnostic tool and should be conducted with and without contrast.
Resection treatment of choice.
Resectability dependent upon site of the tumor with 95% of extremity lesions and 20% of paraspinal lesions able to be resected.
Chemotherapy of no value.
Radiation not adequately tested.
Local recurrence rates 32-65% following local resection.
Median time to local recurrence 5-32 months.