 Gangliosidosis is a group of rare, inherited lysosomal storage disorders caused by the harmful accumulation of fatty substances called gangliosides in the body’s cells.
Gangliosidosis is a group of rare, inherited lysosomal storage disorders caused by the harmful accumulation of fatty substances called gangliosides in the body’s cells.
A group of rare, progressive, neurodegenerative lysosomal storage disorders caused by deficiency of specific enzymes needed to break down gangliosides (glycosphingolipids found primarily in neuronal tissue).
Accumulation of gangiosides occurs, particularly in the brain and spinal cord.
This buildup is caused by a deficiency in specific enzymes needed to break these substances down, leading to progressive neurodegeneration.
Gangliosidoses result from inherited defects in lysosomal hydrolases or lipid-binding proteins that normally degrade gangliosides.
There are two primary forms of the disease, each caused by different enzyme deficiencies:
GM1 Gangliosidosis: Caused by a deficiency of the enzyme beta-galactosidase due to mutations in the GLB1 gene.
In GM1 gangliosidosis, mutations in the GLB1 gene cause β-galactosidase deficiency, leading to accumulation of GM1 ganglioside and related glycoconjugates in lysosomes, particularly in neurons.
It often involves systemic symptoms like an enlarged liver and spleen and skeletal abnormalities.
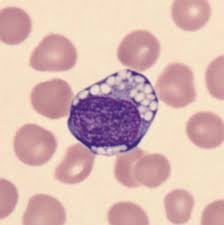
In gangliosidosis, white blood cells (specifically lymphocytes) often exhibit, large, sharply, defying, vacuoles, and abnormal granules (such as eosinophils) during routine blood smears.
Analyzing white blood cells (leukocytes) is a primary diagnostic method to measure deficient lysosomal enzyme
GM2 Gangliosidosis: Caused by a deficiency of the enzyme beta-hexosaminidase: deficiency of hexosaminidase A (Tay-Sachs) or both hexosaminidases A and B (Sandhoff) causes GM2 ganglioside accumulation.
This storage material causes lysosomal swelling, cellular damage, and progressive neurodegeneration.
The most well-known forms are: Tay-Sachs Disease (HexA deficiency) Sandhoff Disease (HexA and HexB deficiency)
Both GM1 and GM2 are typically categorized based on the age when symptoms first appear, which often correlates with the level of residual enzyme activity:
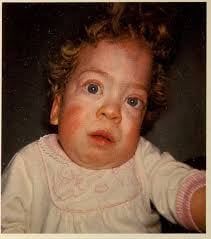
Type I (Infantile) | Birth to 6 months | Hypotonia, startle response, seizures, “cherry-red spot” in the eye.
Onset by 6 months with developmental delay/regression, hypotonia, hepatosplenomegaly, skeletal dysplasia (vertebral beaking, kyphosis), coarse facial features, cherry-red spot on macula, seizures, and profound intellectual disability.
Death typically occurs in early childhood.
Rapidly fatal; usually early childhood.
Type II (Juvenile)
18 months to 5 years-Loss of coordination (ataxia), speech decline, cognitive regression
Mid-childhood to early adulthood
Type III (Adult)
Teens to 30s-Muscle weakness, tremors, speech problems, milder cognitive decline May have a near-normal lifespan
Genetics and Inheritance These conditions are autosomal recessive, meaning a child must inherit a mutated gene from both parents to be affected. If both parents are carriers, there is a 25% chance with each pregnancy that the child will have the disorder.
Management Currently, there is no cure for gangliosidosis.
Treatment is primarily supportive and palliative, focusing on:
Seizure management with anticonvulsants.
Physical and speech therapy to maintain mobility and communication.
Nutritional support, such as feeding tubes for patients with swallowing difficulties.
Experimental therapies like gene therapy, enzyme replacement, and substrate reduction are currently being investigated in clinical trials.
Type II (late infantile/juvenile): Onset at 18 months to 5 years with developmental delays, ataxia, dystonia, dysarthria, progressive brain atrophy, and slower progression than Type I.
Type III (adult): Mildest form with dystonia, gait or speech disturbances, and mild skeletal changes.
GM2 Gangliosidosis:
Similar classification with infantile forms showing cherry-red spot, exaggerated startle response, progressive neurodegeneration, and early death.
Diagnosis involves enzymatic activity assays showing deficient β-galactosidase (GM1) or hexosaminidase (GM2) activity, followed by genetic sequencing to identify pathogenic variants.
In GM1, most mutations cluster in exons 2, 6, 15, and 16 of GLB1, with 261 pathogenic variants described (predominantly missense/nonsense mutations).
Treatment:
No curative treatments existed beyond palliative care.
Gene therapy: AAV9-mediated gene therapy trials are currently recruiting patients with GM1 gangliosidosis.
A 2026 phase 1-2 trial of AAV9-GLB1 in Type II GM1 gangliosidosis showed improved survival, increased CSF enzyme levels, and corrected neuroimaging abnormalities at 3 years.
Substrate reduction therapy: Venglustat (glucosylceramide synthase inhibitor) is being evaluated in Type II and III patients.
Miglustat combined with ketogenic diet showed survival benefit in infantile GM1 in a single study.
Supportive care:
Chest physiotherapy was associated with increased survival in infantile GM1.
Management includes seizure control, feeding support, and multidisciplinary care.
Enzyme replacement therapy, bone marrow/hematopoietic stem cell transplantation, and molecular chaperones have limited efficacy, particularly for CNS manifestations due to blood-brain barrier limitations.
The prognosis remains poor for infantile forms, with death in early childhood, though gene therapy represents a potentially transformative approach for these devastating disorders.