 Friedreich’s ataxia (FA) is an autosomal-recessive genetic disease that causes difficulty walking, a loss of sensation in the arms and legs, and impaired speech that worsens over time.
Friedreich’s ataxia (FA) is an autosomal-recessive genetic disease that causes difficulty walking, a loss of sensation in the arms and legs, and impaired speech that worsens over time.
FRDA affects Indo-European populations.
It is rare in East Asians, sub-Saharan Africans, and Native Americans.
FRDA is the most prevalent inherited ataxia, affecting approximately 1 in 40,000 with European descent.
A 1990–1996 study of Europeans calculated the incidence rate was 2.8:100,000.
The prevalence rate of FRDA in Japan is 1:1,000,000.
Males and females are affected equally.
The estimated carrier prevalence is 1:100.
Also known as spinocerebellar ataxia.
Symptoms generally start between 5 and 20 years of age.
Many patients develop hypertrophic cardiomyopathy.
Patients frequently require a mobility aid such as a cane, walker, or wheelchair in their teens.
With progression of disease some patients may lose sight and hearing.
Other complications include scoliosis and diabetes.
Frequency 1 in 50,000.
The condition is caused by mutations in the FXN gene on chromosome 9, which makes a protein called frataxin.
In FA cells produce less frataxin.
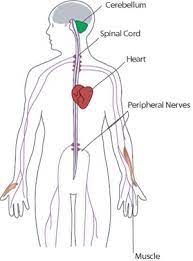
Degeneration of nerve tissue in the spinal cord causes the ataxia.
The sensory neurons essential for directing muscle movement of the arms and legs through connections with the cerebellum, are particularly affected.
The spinal cord becomes thinner, and nerve cells lose some myelin sheath.
No effective treatment is known.
FA shortens life expectancy due to heart disease, but some people can live into their 60s or older.
It affects one in 50,000 people in the United States and is the most common inherited ataxia.
Rates are highest in people of Western European descent.
Symptoms typically start between the ages of 5 and 15.
In late-onset FRDA, they may occur after age 25 years.
The symptoms are broad, but consistently involve gait and limb ataxia, dysarthria and loss of lower limb reflexes.
All individuals with FA develop neurological symptoms, including dysarthria and loss of lower limb reflexes.
More than 90% of patients with FA present with ataxia.
Cardiac issues are very common with early onset FA.
Most patients develop heart problems such as enlargement of the heart, symmetrical hypertrophy, heart murmurs, atrial fibrillation, tachycardia, hypertrophic cardiomyopathy, and conduction defects.
Scoliosis is present in about 60% of FA patients.
7% of people with FA also have diabetes.
Other symptoms include:
36.8% experience decreased visual acuity, which may be progressive and could lead to functional blindness;
Hearing loss is present in about 10.9% of cases, some patients report bladder and bowel symptoms;
Advanced stages of disease are associated with supraventricular tachyarrhythmias, most commonly atrial fibrillation,
Cerebellar effects such as nystagmus, fast saccadic eye movements, dysmetria, truncal ataxia, and stomping gait.
Involvement of the dorsal column may result in loss of vibratory and proprioceptive sensation.
Most patients with FA require mobility aids such as a cane, walker, or wheelchair by early 20s.
The progressive loss of coordination and muscle strength leads to the full-time use of a wheelchair.
It is a progressive process with increased stumbling and frequent falls.
By the third decade there is a loss in the ability to stand or walk without assistance and require a wheelchair for mobility.
Non-neurological symptoms such as scoliosis, pes cavus, cardiomyopathy and diabetes are more frequent amongst the early-onset cases.
In 96% of cases, the mutant FXN gene has 90–1,300 GAA trinucleotide repeat expansions in intron 1 of both alleles.
This expansion causes epigenetic changes and formation of heterochromatin near the repeat, resulting in reduced transcription of the gene and low levels of frataxin.
The length of the shorter GAA repeat is correlated with the age of onset and disease severity.
People with FDRA might have 5-35% of the frataxin protein compared to healthy individuals.
Heterozygous carriers of the mutant FXN gene have 50% lower frataxin levels, but this decrease is not enough to cause symptoms.
4% of cases, the disease are due to a point mutation, with an expansion in one allele and a point mutation in the other.
FA affects the nervous system, heart, pancreas, and other systems.
Degeneration of nerve tissue in the spinal cord causes ataxia.
The sensory neurons essential for directing muscle movement of the arms and legs through connections with the cerebellum are particularly affected.
The disease primarily affects the spinal cord and peripheral nerves.
As the spinal cord becomes thinner, nerve cells lose some myelin sheath.
The spinal cord diameter is smaller than in unaffected individuals, due to smaller dorsal root ganglia.
In FA the motor neurons of the spinal cord are affected to a lesser extent than sensory neurons.
In peripheral nerves there is a loss of large myelinated sensory fibers.
The dentate nucleus of the cerebellum is also affected by FA.
The heart often develops some fibrosis with FA, with the development oof left-ventricle hypertrophy and dilatation of the left ventricle.
Frataxin assists iron-sulfur protein synthesis in the electron transport chain to generate adenosine triphosphate.
Adenosine triphosphate is the energy molecule necessary to carry out metabolic functions in cells.
Adenosine triphosphate also regulates iron transfer in the mitochondria by providing a proper amount of reactive oxygen species (ROS) to maintain normal processes.
One result of frataxin deficiency is mitochondrial iron overload, which damages many proteins due to effects on cellular metabolism.
Without frataxin, the energy in the mitochondria falls, and excess iron creates extra ROS, leading to further cell damage.
Diagnosis:
Balance difficulty, loss of proprioception, an absence of reflexes, and signs of other neurological problems are common signs from a physical examination.
Diagnostic tests confirming physical examination findings: electromyogram, nerve conduction studies, electrocardiogram, echocardiogram, blood tests for elevated glucose levels and vitamin E levels, and scans such as X-ray radiograph for scoliosis.
MRI and CT scans of brain and spinal cord are done to rule out other neurological conditions.
A genetic test is conducted to confirm the diagnosis.
Differential diagnoses: Charcot-Marie-Tooth types 1 and 2, ataxia with vitamin E deficiency, ataxia-oculomotor apraxia types 1 and 2, and other early-onset ataxias.
Management:
No cure exists.
Medications may be prescribed to improve spasticity, abnormal movements and heart disease.
Physical therapists play a critical role: correct posture, muscle use, and the identification and avoidance of features that aggravate spasticities.
Physical therapy typically includes intensive motor coordination, balance, low intensity strengthening exercises, stretching and muscle relaxation exercises, transfer and locomotion independence, muscle strengthening, increased physical resilience, “safe fall” strategy, learning to use mobility aids, learning how to reduce the body’s energy expenditure, developing specific breathing patterns, and speech therapy.
Orthoses can promote correct posture, support normal joint alignment, stabilize joints during walking, improve range of motion and gait, reduce spasticity, and prevent foot deformities and scoliosis.
Functional electrical stimulation or transcutaneous nerve stimulation devices may alleviate symptoms.
Assistive devices such as a cane, walker, or wheelchair may be required for mobility and independence.
Cardiac abnormalities can be controlled with ACE inhibitors, sometimes used in conjunction with beta blockers and other agents for heart failure.
An automated implantable cardioverter-defibrillator can be implanted after a severe heart failure.
Surgery may correct deformities caused by abnormal muscle tone:
Titanium screws and rods inserted in the spine help prevent or slow the progression of scoliosis.
Surgery to lengthen the Achilles tendon can improve independence and mobility to alleviate equinus deformity.
Patients diagnosed at a younger age or with longer GAA triplet expansions tend to have more severe symptoms.
Congestive heart failure and abnormal heart rhythms are the leading causes of death, but people with fewer symptoms can live into their 60s or older.
FRDA follows the same pattern as haplogroup R1b.
Haplogroup R1b is the most frequently occurring paternal lineage in Western Europe.
FRDA and Haplogroup R1b are more common in northern Spain, Ireland, and France, rare in Russia and Scandinavia, and follow a gradient through central and eastern Europe.