 A ciliopathy is any genetic disorder that affects the cellular cilia or the cilia anchoring structures, the basal bodies,or ciliary function.
A ciliopathy is any genetic disorder that affects the cellular cilia or the cilia anchoring structures, the basal bodies,or ciliary function.
Ciliopathy disorders that affect the function or structure of cilia, which are hair-like structures found on the surface of many types of cells in the human body.
Cilia have important roles in various biological processes, including the movement of fluids across cell surfaces, the detection of signals in the environment, and the development and function of organs.
Primary cilia play important roles in chemosensation, mechanosensation, and thermosensation.
A number of common observable characteristics of mammalian genetic disorders and diseases are caused by ciliary dysgenesis and dysfunction.
Cilia are implicated in a wide variety of human genetic diseases by numerous proteins involved in mammalian disease localized to the basal bodies and cilia.
Cilia coordinate a large number of cellular signaling pathways, sometimes coupling the signaling to ciliary motility or alternatively to cell division and differentiation.
Ciliopathies can be caused by mutations in genes that are involved in the formation, maintenance, or function of cilia.
These mutations can lead to a wide range of symptoms and health disorders, depending on the specific type of ciliopathy and which organs are affected.
Ciliopathies include:polycystic kidney disease, Bardet-Biedl syndrome, primary ciliary dyskinesia, and retinal degeneration.
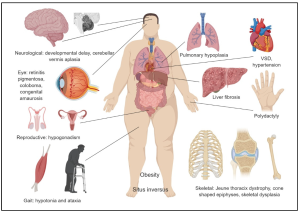
Symptoms of ciliopathies can include: kidney or liver disease, vision and hearing problems, breathing difficulties, developmental delays, and skeletal abnormalities.
There is currently no cure for ciliopathies.
Treatment options focus on managing symptoms and preventing complications.
Abnormal ciliary function while an embryo is developing can lead to a set of malformations that can occur regardless of the particular genetic problem.
Ciliopathies are a clustering of a set of characteristic physiological features which define whether a syndrome is a ciliopathy.
Ciliopathies are usually involve proteins that localize to motile and/or immotile primary cilia or centrosomes.
It affect ciliary function through proteolytic cleavage of ciliary proteins.
A wide variety of symptoms and potential clinical features of ciliopathy:
Dandy–Walker malformation (cerebellar vermis hypoplasia, usually with hydrocephalus)
Agenesis of the corpus callosum
Situs inversus
Posterior encephalocele
Polycystic kidneys
Postaxial polydactyly
Liver disease
Retinitis pigmentosa
Intellectual disability
Phenotypes sometimes associated with ciliopathies can include:
Anencephaly
Breathing abnormalities
Cerebellar vermis hypoplasia
Diabetes
Exencephaly
Eye movement abnormalities
Hydrocephalus
Hypoplasia of the corpus callosum
Hypotonia
Infertility
Cognitive impairment/defects
Obesity
Polydactyly
Respiratory dysfunction
Renal cystic disease
Retinal degeneration
Sensorineural deafness
Spina bifida
The motile cilium is a nanomachine composed of perhaps over 600 proteins in molecular complexes, many of which also function independently.
Cilia function as mechano- or chemosensors and as a cellular global positioning system to detect changes in the surrounding environment.
Ciliary signaling plays a role in the initiation of cellular replacement after cell damage.
Cilia play sensory role mediating specific signaling cues, and a secretory role in which a soluble protein is released to have an effect downstream of the fluid flow in epithelial cells, and can mediate fluid flow directly in the case of motile cilia..
Primary cilia in the retina play a role in transferring nourishment to the non-vascularized rod and cone cells from the pigment epithelial vascularized cells several micrometres behind the surface of the retina.
Signal transduction pathways involved with cilia include the Hedgehog signaling pathway and the Wnt signaling pathway.
Dysfunctional ciliated epithelial cellular dysfunction. can lead to:
Chemosensation abnormalities
Defective thermosensation or mechanosensation
Cellular motility dysfunction
Issues with displacement of extracellular fluid
Paracrine signal transduction abnormalities
In organisms of normal health, cilia are critical for:
development
homeostasis
reproduction
In two of the diseases caused by malfunctioning cilia, Meckel–Gruber syndrome and Bardet–Biedl syndrome, patients who carry mutations in genes associated with both diseases have unique symptoms that are not seen in either condition alone.
The genes linked to the two different conditions interact with each other during development.
A particular phenotype can overlap, with several conditions in which primary cilia are also implicated in pathogenicity.
List of ciliopathies:
Alström syndrome
Asphyxiating thoracic dysplasia (Jeune syndrome
Bardet–Biedl syndrome
Ellis–van Creveld syndrome
Joubert syndrome
Brain
Leber congenital amaurosis
McKusick–Kaufman syndrome
Meckel–Gruber syndrome
Nephronophthisis
Orofaciodigital syndrome
Polycystic kidney disease
Primary ciliary dyskinesia (Kartagener syndrome)
Senior–Løken syndrome-eye
Sensenbrenner syndrome (cranioectodermal dysplasia)
Short rib–polydactyly syndrome
Likely ciliopathies:
Acrocallosal syndrome
Acromelic frontonasal dysostosis
Arima syndrome
Biemond syndrome
COACH syndrome
Conorenal syndrome
Greig cephalopolysyndactyly syndrome
Hydrolethalus syndrome
Johanson–Blizzard syndrome
Mohr syndrome (oral-facial-digital syndrome type 2)
Neu–Laxova syndrome
Opitz G/BBB syndrome
Pallister–Hall syndrome
Papillorenal syndrome
Renal–hepatic–pancreatic dysplasia
Varadi–Papp syndrome (oral-facial-digital syndrome type
There are many other possible ciliopathies.