Rare process with a prevalence of 10 cases per million people.
Due to pituitary tumors arising in somatotroph cells with aberrant secretion of growth hormone.
High growth hormone and insulin-like growth factor 1 levels are associated with somatic and metabolic dysfunction.
Acromegaly is the result of excess growth hormone secretion by generally benign adenoma arising in adult pituitary somatotrophic.
The incidence of acromegaly is approximately 10 cases per 1 million persons.
Roughly 20% of all pituitary tumors secrete growth hormone, and 95% appear to be sporadic without a known genetic cause.
Approximately 70% of patients with acromegaly have invasive macroadenoma at diagnosis.
It may be associated with germline AIP mutations, the McCune-Albright syndrome, or acidophilic stem cell adenomas.
Clinical findings may include: frontal bossing, prognathism, deep nasolabial folds, thickened skull, widened spacing in teeth, facial edema, underbite, macroglossia, enlargement of feet, hands, excessive sweating, acne, headache, diabetes, thickened skin, skin tags, tall stature, insulin resistance, hypogonadism, hypertension, sleep apnea, arthralgia, carpal tunnel syndrome, colon polyps, and cardiac valve dysfunction.
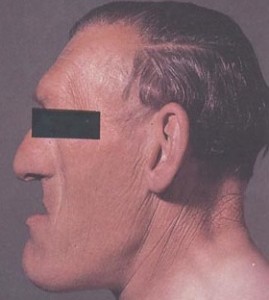
Acromegaly is characterized by facial and cranial changes, such as frontal bossing, large nose and lips, increased dental spacing, and prognathism, and acral enlargement in 95%, headache in 60% of patients, arthropathy in 70% of patients, hyperhidrosis in 65% of patients and compressed or collapsed vertebrae in 50% of patients, diabetes, or pre-diabetes in 50% of patients, hypertension in 35%, cardiomyopathy leading to congestive heart failure in 10%, valvular heart disease up to 30%, sleep apnea 65%, carpal tunnel syndrome 64% and hyperprolactinemia 30%.
Dual x-ray absorptiometry can detect specific acromegaly related lipodystrophy with reduced visceral adipose tissue and ectopic lipid deposition in muscle.
Coexisting conditions include headache and acral and soft tissue changes.
Headaches in acromegaly appear to be due to stretching of the dura mater and invasion of pain producing structures.
Patients may present with unusual headache phenotypes such as cluster headaches with unilateral short-lasting neuralgiform headache attacks with conjunctival injection and tearing.
Slowly progressive enlargement of the face and extremities associated with cardiac, rheumatologic, restpiratory, and metabolic abnormalities tied to elevations in circulating growth hormone and insulin like growth factor-1.
Prognathism leads to incisor separation and jaw malocclusion.
Obstructive sleep apnea and excessive snoring are hallmarks of uncontrolled acromegaly.
Arthropathy is reported in about 70% of patients with polyarticular arthritis, osteophytosis, dorsal kyphosis, and vertebral fractures.
Associated with structural changes including: thickening of the heart muscle, dilatation of the ventricles, diminished diastolic function, hypertension, arrhythmias. left ventricular dysfunction, and an increase the aortic root diameter.
Not unusual for patients with acromegaly to have hypertension, valve dysfunctions of mainly aortic and mitral, and tricuspid valves and arrhythmias.
A major contributor to death from cardiovascular causes in acromegaly is hypertension, with the prevalence ranging from 30 to 60%.
Structural changes are compounded by common comorbidities which include: hypertension, dyslipidemia, insulin resistance, and glucose intolerance with increased risk of diabetes.
About 30% of patients with acromegaly have high prolactin levels, often with galactorrhea.
Colon polyps were detected in 32% of patients.
Acromegaly is associated with increased incidence of cancer: colorectal, kidney, and thyroid: not confirmed in other studies.
Colonic polyps, particularly multiple, or polyps in the left colon as well as advanced adenoma and colorectal carcinoma are more common in patients with acromegaly.
Higher death rates associated with acromegaly due to cardiovascular, respiratory and cerebrovascular disorders and also possibly cancer.
Somatotroph cells originate in the anterior pituitary gland producing an excess of growth hormone.
Somatotroph cell development and proliferation related to the Prophet of Pit-1 (PROP1) gene which controls embryonic development of cells of the Pit-1 transcription factor lineage.
Growth hormone secreted as a 191 amino acid, 4-helix bundle protein and a less abundant 176 amino acid form which enter the circulation under the control of the hypothalamic releasing and hypothalamic inhibiting hormones that traverse the hypophysial portal system by influencing specific somatotrophic surface receptors.
Release of growth hormone is pulsatile in nature.
Growth hormone releasing hormone induces synthesis and secretion of the hormone while somatostatin suppresses its secretion.
Finding of elevated serum level of insulin like growth factor (IGF-1) is suggestive of acromegaly.
Diagnosis can be delayed by approximately 10 years after the onset of symptoms as patients first seek dental, orthopedic, rheumatologic, or cardiac care.
Approximately 20% of patients seekcare because of altered facial appearance, enlarge the extremities, or both.
Diagnosis confirmed by lack of growth hormone suppression after glucose suppression testing.
Binding of growth hormone to hepatic receptors leads to systemic release of IGF-1.
Growth hormone regulated by ghrelin.
Affects the entire body with risk of substantial morbidity and premature mortality if not recognized and treated.
Up to 70% of patients, have large joint pain and axial arthropathy with progressive functional disability and impaired quality of life.
Imaging findings with MRI show extensive joint cartilage, increased water content, and small cysts and marrow lesions.
Patients may have vertebral fractures with normal or slightly reduced bone mineral density, and patients may have bilateral or relapsing carpal tunnel syndrome.
Diagnosis delay averages 5-10 years, with alternative diagnoses of diabetes, hypertension, osteoarthritis, carpal tunnel syndrome, joint replacement, sleep apnea, malocclusion and renal stones being made before the appropriate diagnosis.
Sleep apnea and disordered breathing is detected with polysomnography in up to 80% of patients with newly diagnosed acromegaly: caused by obstruction in acromegaly associated cranial facial anomalies, macroglossia, and laryngeal wall thickening, which may also lead to heavy snoring.
Patients may have typical skin manifestations of multiple skin tags, acne, oily and thicken skin and acanthosis.
Findings and women include menstrual disturbances, uterine leiomyomas, ovarian cysts, and infertility.
Hypopituitarism and hypogonadism may occur at approximately half the patients as the result of mass effect of the tumor, hyperprolactinemia, or both.
Coexisting conditions, roughly for per patient, as well as increased mortality, are directly correlated with delayed diagnosis, and are more common in older patients, particularly women.
The earliest systemic manifestations before diagnosis are hypertension, and carpal tunnel syndrome.
Vertebral fractures detected in 30% of patients at presentation are strongly associated with the diagnostic delay.
Hypertension results from sodium returning affects of growth hormone, inhibition in atrial natriuretic peptide by insulin like growth factor-1, and increased peripheral vascular resistance triggered by both hormones.
Excess growth hormone increases production of pro inflammatory mediators that degrade structural elements of the aortic and mitral valve, precipitating regurgitation .
Associated with progressive myocardial fibrosis and involvement of the conduction system of the heart increase the risk for arrhythmias and conduction disorders.
Altered hormonal activity changes In the sympathoadrenalmedullary pathway, promotes endothelial dysfunction, valvular disease and cardiomyopathy.
In normal persons growth hormone levels are usually undetectable through most of the day but there are approximately 10 intermittent pulses of growth hormone per 24 hour, most often at night when the level can be as high as 30 microgm per liter.
Peaks of growth hormones may overlap with elevated levels as seen in acromegaly.
Fasting increases secretion of growth hormone, while aging and obesity decrease bursts of the hormone.
Fasting or random growth hormone level results may be normal because of the pulsatile secretion of the hormone and the impracticality of measuring pulsatile levels.
Definitive testing for the diagnosis is the use of the oral glucose tolerance test and measurement of growth hormone every 30 minutes for 2 hours.
Best screening test measurement of serum insulin like growth factor 1 (IGF-1) level.
IGF-1 elevated levels for the patient’s age are highly specific for acromegaly and correlate with disease activity.
Because of the pulsatile nature of adenoma growth hormone secretion reliance on random measurement for diagnosis of IGF-1 is precluded.
IGF-1 an integrated measure of overall growth hormone secretion.
IGF-1 varies with age and decreases with age.
IGF-1 falsely elevated in pregnancy as placental production of a growth hormone variant that stimulates production of IGF-1.
Diagnosis of acromegaly is established by using an ultra sensitive assay to document nadir growth hormone levels of more than 0.4 µg per liter during a 75 g glucose load.
Measurement of growth hormone levels after a glucose load, global pituitary function tests, and pituitary MRI imaging with gadolinium are carried out to complete diagnostic processes.
TREATMENT:
Treatment goals include ablating or controlling the pituitary mass, suppressing growth hormone and IGF-1 hypersecretion, and preventing the development of associated disorders while maintaining anterior pituitary function, normalization of growth, hormone levels during an oral glucose tolerance test, prevention of associated conditions, preservation of anterior pituitary function, and normalization of life expectancy through surgery, medical treatment, and radiation therapy.
Medical therapy is generally reserved for those with persistent or recurrent disease after pituitary surgery.
Surgical resection resulted in control of growth hormone secretion and IGF-1 levels in 75% of patients with micro adenomas AND 61% of patients with macroadenomas.
In patients with macroadenoma invading the cavernous sinus show persistent growth hormone hypersecretion after surgery, so it is not recommended his first line therapy unless vital structures are endangered.
Upper airway obstruction, poorly controlled, diabetes, severe, hypertension, or heart failure, if present should be managed medically, which may require a delay in surgery.
Readmission after pituitary surgery is slightly higher than 2%, though postoperative deficiencies of vasopressin, gonadotropins, and thyrotropin are sporadically observed.
Surgical removal of at least 75% of a growth hormone macroadenoma may enhance the efficacy of subsequent treatment with somatostatin receptor ligands.
Radiosurgery shows biochemical remission it up to 59% of patients, with the meantime to remission of 38 months and the meantime to recurrence of 17 months.
Somatostatin receptor ligands octreotide and lanreotide inhibit growth hormone secretion and their use is associated with an overall control of growth hormone secretion and of IGF-1 levels in 55% of patients.
Somatostatin receptor ligands result in about a 53% reduction in tumor mass.
Octeotide long-acting repeatable, and lanreotide autogel are the available.
Long acting somatostatin receptor ligands with monthly injections of octreotide or deep subcutaneous injections of lanreotide maintaine biochemical control in 30 to 50% of patients, and higher estimates for efficacy are generally reported in studies involving patients who are pre-selected for their positive response to short acting octreotide.
With the use of the agents, soft tissue swelling and headache usually resolve, sleep apnea abates, and left ventricular function improves, but hypertension may persist.
Somatostatin receptor ligands may induce tumor shrinkage by 20% or more in around 50% of patients, as well as safely improving the clinical picture.
Pasitreotide is a long acting release somatic statin receptor ligand given monthly and is more effective and normalizing the hormonal profile then octreotide however it frequently causes hyperglycemia and is currently indicated as a second line treatment.
Oral ocreotide capsules and other oral SSTR binders are available for the management of acromegaly.
Pegvisomant a pegylated growth hormone analogue with amino acid substitutions, resulting in a functional block of growth hormone receptor and hepatic production of IGF –1, leads to biochemical control in approximately 90% of patients.
The readmission rate after pituitary surgery is slightly higher than 2%, although postoperative deficiencies and vasopressin, gonadotropins, and thyrotropin were sporadically observed.
Surgical removal of at least 75% of a growth hormone of macroadenoma may enhance the efficiency of subsequent treatments with somatostatin receptor Lyons.
Dopamine agonist for a patient with mild disease may normalize IGF-one levels in some patients with disease that is resistant to somatostatin therapy.
Pegvisomant is a growth hormone receptor antagonist that blocks peripheral growth hormone action and subsequent IGF-1 production is useful for patients with resistant disease to somatostatin receptor ligands as well as in patients with hyperglycemia: the drug enhances insulin sensitivity.
Pegvisomant added to Pituitary directed somatostatin receptor ligand offers greater efficacy then either drug alone.
Pegvisomant and pasireotide are associated with high likelihood of effectivity in the most effective single agents.
In patients treated for acromegaly all cause mortality is not increased above the general population in patients who achieve a normal serum IGF –1 level.