 A genetic disorder that inhibits endochondral ossification, resulting in disproportionate short stature and clinically significant medical complications.
A genetic disorder that inhibits endochondral ossification, resulting in disproportionate short stature and clinically significant medical complications.
The most common form of disproportionate short stature.
Prevalence of one in 25,000 live births.
A genetic disorder that results in dwarfism.
Frequency is 1 in 27,500 people.
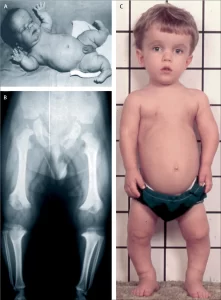
It manifests with short arms and legs, while the torso is typically of normal length.
Characterized by disproportionate short statue with relatively short limbs, trident hands, mid face hypoplasia, and foramen magnum stenosis.
Shortening of the proximal limbs is called rhizomelic shortening.
Individuals affected have an average adult height of 4 ft 4 inches for males and 4 ft for females.
Patients also have an enlarged head and prominent forehead.
Mental status is normal.
Possible complications include:
Ear infections, lordosis, back pain, spinal stenosis, hydrocephalus, dental malocclusion, repeated otitis media can be observed.
The risk of death in infancy is increased due to the likelihood of compression of the spinal cord with or without upper airway obstruction.
It is caused by a mutation in the FGFR3 gene.
The FGFR3 gene is mainly responsible for making the protein, fibroblast growth factor receptor.
Fibroblast growth factor receptor protein contributes to the production of collagen and other structural components in tissues and bones.
When the FGFR3 gene is mutated it interferes with how this protein interacts with growth factors leading to complications with bone production.
FGFR3 is a negative regulator of chondrocyte proliferation and differentiation in the growth plate.
FGFR3 variants result in excessive activation of the FGFR3 protein and subsequently increased intracellular signaling leading to the inhibition of chondrocyte assembling and proliferation in the growth plates.
The FGFR3 gene mutation causes cartilage not able to be fully develop into bone, causing disproportionately shorter height.
This inhibition occurs via the signal transducer and activator of transcription 1 (STAT 1) and mitogen activated protein kinase (MAPK) pathways in chondrocytes.
About 80% of cases this occurs as a new mutation during early development.
A new gene mutation for achondroplasia is exclusively inherited from the father and occurs during spermatogenesis.
One mutant copy of the FGFR3 gene being sufficient to cause achondroplasia, while two copies of the mutant gene are invariably fatal.
With achondroplasia there is a 50% chance of passing dwarfism to each of their children.
When both parents have achondroplasia there is a 50% chance the child will have achondroplasia, 25% chance the child will not, and a 25% chance that the child will inherit the gene from both parents resulting in double dominance and leading to death.
The remaining cases result from inheritance from one’s parents in an autosomal dominant manner.
The diagnosis can be made by fetal ultrasound with discordance between the femur length and biparietal diameter by age.
In patients with two affected genes, survival is unlikely.
Associated with 10 year shorter life expectancy.
Differential diagnosis:
Hypochondroplasia, thanatophoric dysplasia, cartilage-hair hypoplasia, pseudoachondroplasia.
Diagnosis is usually established by clinical findings.
Findings include: Short fingers and toes with trident hands, large head with prominent forehead and frontal bossing, small midface with a flattened nasal bridge, spinal kyphosis or lordosis, varus (bowleg) or valgus (knock knee) deformities, frequent ear infections, sleep apnea and hydrocephalus.
Radiologic findings :
The skull is large,
A narrow foramen magnum.
Relatively small skull base.
Short and flattened vertebral bodies.
Relatively large intervertebral disk height.
Congenitally narrowed spinal canal.
Iliac wings are small and squared.
Tubular bones are short and thick with metaphyseal cupping and flaring and irregular growth plates.
Fibular overgrowth.
The hand is broad with short metacarpals and phalanges.
The ribs are short with cupped anterior ends.
Patients with achondroplasia are often “double jointedâ€
Thoracolumbar gibbus in infancy.
Treatment efforts include:
to treat or prevent obesity, hydrocephalus, obstructive sleep apnea, middle ear infections, or spinal stenosis.
The frequency of mutations in sperm leading to achondroplasia increase with paternal age, and exposure to ionizing radiation.
Achondroplasia in the children of fathers over 50 years of age is 1 in 1875 compared to 1 in 15,000 in the general population.
It can be detected before birth by prenatal ultrasound.
A DNA test can be performed before birth.
No known cure exists.
Human growth hormone does not help people with achondroplasia.
After the second year of growth hormone therapy, beneficial bone growth decreases.
Often children have less muscle tone, and commonly have delayed walking and motor skills, bowed legs, scoliosis, lordosis, arthritis, impaired joint flexibility, breathing problems, ear infections, crowded teeth, and hydrocephalus.
Adults are obese and have sleep apnea.
Adults commonly suffer from numbness or tingling in their legs because of nerve compression.
Pregnancy is associated with considered higher risk, and pregnant women with achondroplasia generally have their babies delivered through C-sections to prevent complications that could occur with a natural birth.
Associated with increased mortality from birth to four years of age, and also in the fourth and fifth decades of life.
Caused by an autosomal dominant mutation in the fibroblast growth factor receptor 3 gene (FGFR 3) that activates the mitogin-activated protein kinase (MAPK) extracellular signal-regulated kinase pathway in chondrocytes, which inhibits endochondral ossification.
Hallmark features include short stature with rhizomelic limb shortening and macrocephaly.
Medical complications include: hydrocephalus, hypotonia, back pain, leg pain, conductive hearing loss, and speech delay.
Maybe associated with tonsillar hypertrophy resulting in obstructive of sleep apnea and respiratory insufficiency.
Foraminal magnum stenosis and cervicomedullary compression may result in central apnea which may increase the risk of sudden death in infancy.
Associated with developmental milestones impairments, functional limitations affecting quality-of-life and chronic pain, all of which can lead to psychosocial challenges.
In children with achondroplasia a once daily subcutaneous administration of Vosoritide was associated with sustained increasing annualized growth velocity for up to 42 months.
Long-term study of growth hormone therapy resulted in an increase in final height of 3.5 cm in male patients in 2.8 cm in female patients.
Limb-lengthening surgery can improve height but does not prevent other medical complications.
Vosoritide is a biologic analog of C-type natriuretic peptide, a potent stimulator of endochondral ossification.
Once daily s.c. Vosoritide results in a sustained increase in annualized growth velocity for about 42 months.
Infigratinib an oral bioavailable FGFRI-3 selective tyrosine kinase inhibitor increased the annualized height velocity, with a decrease in the ratio of the upper to lower body segment at 18 months, reflecting the predominant growth of the limbs rather than the trunk.
Infigratinib is more effective than vosoritide in growth velocity.