 Life threatening disorder that is treatable by timely therapy.
Life threatening disorder that is treatable by timely therapy.
Prevalence 120 per 1 million.
Presentation usually nonspecific: patients commonly experience fatigue, nausea, vomiting, abdominal pain, muscle or joint pain, anorexia and weight loss.
Also known as Addison’s disease.
Slightly higher prevalence among women compared with men.
20% of patients report nonspecific symptoms for more than five years prior to diagnosis.
Diagnosis: in the absence of a serious illness, early morning testing of cortisol, corticotropin, and dehydroepiandrosterone sulfate usually provides sufficient evidence to diagnose adrenal insufficiency
Addison’s disease is an endocrine disease that results from hypocortisolism caused by adrenal gland insufficiency.
Adrenal insufficiency is significant because it is correlated with decreased ability to maintain blood pressure and blood sugar, a defect that can prove to be fatal.
Adrenocortical production of cortisol is regulated by the hypothalamic-pituitary-adrenal axis.
The hypothalamus produces CRH (cortical releasing hormone) in response to light, exhibiting diurnal variation and stressors, such as fever, hypoglycemia, hypotension, pain, and/or acute illness.
CRH induces formation and release of corticotropin from the anterior pituitary, which stimulates the adrenal cortex to produce circulating glucocorticoids, mineralcorticoids, and androgens.
Primary adrenal insufficiency is characterized by deficient production of cortisol and aldosterone, by the adrenal gland and is most commonly caused by autoimmune adrenalitis.
Primary adrenal insufficiency results in deficiency of all adrenocortical hormones, including low levels of cortisol, aldosterone, and the adrenal androgens dehydroepiandrsterone, and androstenedione as well as marked elevations in corticotropin as a result of decreased negative feedback.
The most common causes of primary adrenal insufficiency are autoimmune adrenalitis, and inherited disorders of the adrenal steroidgenesis, such as congenital adrenal hyperplasia due to 21-hydroxylase deficiency (approximately one and 15,000 live births).
More than 90% of patients with primary adrenal insufficiency have circulating 21-hydroxylase antibodies, which confirms the diagnosis of autoimmune adrenalitis.
Infectious adrenalitis may be the leading cause of primary adrenal insufficiency in areas of the world with high rates of tuberculosis and or HIV infection.
Primary adrenal insufficiency can occur at any age, however, most cases occur between ages 20 and 50 years.
Secondary adrenal insufficiency is characterized by decreased pituitary gland production of corticotropin (adrenocorticotropic hormone, ACTH)) may be caused by pituitary tumors, hemorrhage, inflammation, surgery, or radiation or by medications that suppress corticotropin production, such as opioids.
Primary adrenal insufficiency global prevalence estimates range from 4 to 221 cases per 1 million and secondary adrenal insufficiency occurs globally in 140 to 279 per 1 million individuals.
Glucocorticoid induced adrenal insufficiency (tertiary adrenal insufficiency) is caused by suppression is caused by suppression of corticotropin releasing hormone from the hypothalamus which prevents corticotropin release from the pituitary.
Glucocorticoid induced adrenal insufficiency is the most common form of adrenal insufficiency as up to 1 to 3% of the adult population is prescribed glucocorticoid therapy.
The use of exogenous glucocorticoids in supraphysiological doses of 5 mg of prednisone or greater induces negative feedback to suppress corticotropin releasing hormone and corticotropin production, resulting in decreased endigenous, adrenal production of cortisol and androgens.
With prolonged exposure to supraphysiological doses of glucose cords, atrophy occurs in both the pituitary corticotrophs that produce corticotropin and the adrenal cortex.
Immune checkpoint inhibitor therapies can cause irreversible primary or secondary adrenaline sufficiency with most individuals having secondary adrenal insufficiency from hypophysitis.
Immune checkpoint inhibitor induced insufficiently typically occurs 2 months to eight months after initiation of treatment: approximately 5% of patients have primary adrenal insufficiency due to autoimmune adrenalitis.
Addison’s disease results from damage to the adrenal cortex.
An endocrine or hormonal disorder that occurs in all age groups and afflicts men and women equally.
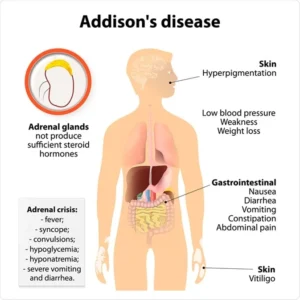
Characterized by weight loss, muscle weakness, fatigue, low blood pressure, and sometimes darkening of the skin in both exposed and nonexposed parts of the body.
Most patients with primary adrenal insufficiency develop hyper pigmentation.
When combined with early morning, cortisol measurement, corticotropin concentrations help determine the type of adrenal insufficiency: nearly all patients with primary adrenal insufficiency have an elevated plasma corticotropin usually greater than 100 pg/mL.
Patients with secondary and glucocorticoid induced adrenal insufficiency have cortical children concentrations that are below, or at the lower end of the reference range, usually less than 20 pg/mL.
In primary adrenal insufficiency all adrenal cortical steroid levels are low, including DHEA.
Dynamic assessment of adrenal function, using corticotropin stimulation evaluates cortisol before in 60 minutes after administration of 250 µg of cosyntropin.
The estimated peak cortisol level following administration of cosyntropin is 18 µg per/dL or greater at 60 minutes.
Other dynamic diagnostic tests include the overnight metyrapone test, insulin induced hypoglycemia, and glucagon stimulation test, which are reserved for patients considered likely to have secondary adrenal insufficiency, but who have a normal or equivocal cosyntropin stimulation test.
In secondary and glucocorticoid induced adrenal insufficiency the insufficiency or deficiency of corticotropin and results in low or low normal levels of DHEAS.
DHEAS serves as an accurate predictive marker of hypothalamic pituitary adrenal axis impairment.
Most patients with adrenal insufficiency have months to years of nonspecific symptoms, including fatigue, nausea, abdominal pain, and anorexia before diagnosis is made.
Some patients (12–19%) report salt cravings and have orthostatic hypotension due to concomitant mineral corticoid deficiency, which may cause hyperkalemia and hyponatremia.
Patients with a glucocorticoid induced adrenal insufficiency may initially develop signs and symptoms of Cushing’s disease-weight gain, central obesity, sarcopenia with proximal muscle weakness, skin fragility, easy bruising, due to superphysiological doses of glucocoerticoids.
These patients develop symptoms of adrenal insufficiency when their dose of glucocorticoids is reduced below physiological supplemental doses of 15 to 25 mg daily of hydrocortisone or 3 to 5 mg daily of prednisone.
Up to 50% of patients with adrenal insufficiency experience an adrenal crisis, which is a life-threatening state of cortical deficiency that can lead to hypovolemic or distributive shock resistance to vasopressors, severe hyponatremia with altered mental status and death.
Adrenal crisis has a mortality rate of 0.5%.
Adrenal crisis is reported by 5 to 8% of patients, and up to 50% of patients with primary adrenal insufficiency experience at least one adrenal crisis prior to diagnosis of adrenal insufficiency.
Adrenal crisis is the cause of death and 15% of patients with Addison’s disease.
Damage to the adrenal cortex may be immune mediated, related to infections with tuberculosis, HIV, fungal infections, hemorrhage, malignancy and use of anticoagulants.
Associated with hypotension, diarrhea, tachycardia, darkening of the skin, extreme weakness, loss of appetite, buccal mucosal lesions, nausea and vomiting, salt craving, slow movement and unintentional weight loss.
Laboratory tests that may be abnormal include: elevated potassium, low sodium, and low cortisol levels.
Treatment:
Management involves determining the appropriate physiological supplemental dose of glucocorticoid, and mineral corticoid, if applicable, patient, education, and management of glucocorticoid therapy during illness or stress to prevent adrenal crisis.
Physiological glucocorticoid therapy is initiated on all patients with adrenal insufficiency and dosing Mike, the amount and timing of the body’s natural production of cortisol.
Hydrocortisone, bioidentical to cortisol, is a short acting oral glucocorticoid at a dose of 15 to 25 mg daily and is sufficient for most adult patients with adrenal insufficiency.
Prednisone or prednisolone is a longer acting oral glucocorticoid and can be administered once daily typically at doses of 3 to 5 mg.
Outcomes of treatment for adrenal insufficiency are equal for hydrocortisone, prednisolone, or prednisone.
Other glucocorticoids, such as methylprednisolone or dexamethasone are not recommended for daily treatment given their long half-life and risk of inducing Cushing syndrome.
Hydrocortisone which is bioidentical to cortisol is a short acting oral Gooogle cord cord with P concentrations 2 to 3 hours after the injection and a dose of approximately 15 to 25 mg daily is generally sufficient for adrenal insufficiency.
Prednisone or prednisolone is a longer acting oral glucocorticoid that is commonly used and can be administered once daily, typically a doses of 3 to 5 mg.
The daily dose can be divided into two doses.
Treatment with replacement corticosteroids will control the symptoms of this disease.
Corticosteroids usually need to taken for life.
Steroid doses are individualized and determined by a patient’s general well-being and function, with the goal of using the lowest steroid dose to achieve subjective well-being.
All patients should receive higher doses of glucocorticoids during stressors, such as infections and surgeries or at times with mental or emotional distress.
People often receive a combination of glucocorticoids (cortisone or hydrocortisone) and mineralocorticoids (fludrocortisone).
Refers to decreased cortisol production and is confirmed by biochemical testing.
Aldosterone production is low in primary adrenal insufficiency and normal in secondary disease: Aldo production by the adrenal gland remains normal in secondary journal insufficiency because it is regulated by angiotensin II and potassium.
During critical illness an increase in circulating and tissue corticosteroids levels are required for adaptive responses.
With severe illness and stress the hypothalamic-pituitary-adrenal (HPA) axis is stimulated, with release of corticotropin releasing hormone (CRH) from the hypothalamus.
Corticotropin releasing hormone stimulates the anterior pituitary to release adrenocorticotropic hormone (ACTH), with ACTH causing increased cortisol production by the adrenal cortex zona fasciculata.
Pigmentary changes often seen in hands and other skin folds is related to coexpression of melanocyte stimulating hormone and corticotropin secreted from the pituitary gland
In acute illness like sepsis, trauma, and burns cortisol production by the adrenal gland is increased by as many as six fold.
Normally bound to corticosteroid-binding globulin with less than 10% free cortisol bioavailable form.
During acute illness cortisol-binding globulin levels decrease by 50% and unbound cortisol levels increases.
Secondary adrenal insufficiency occurs when the pituitary gland fails to produce an adequate amount of corticotropin, a hormone that stimulates the adrenal glands to produce cortisol.
Secondary adrenal insufficiency is much more common than Addison’s disease.
Most patients with secondary and glucocorticoid induced adrenal insufficiency do not develop mineral corticoid deficiency, while most patients with primary adrenal insufficiency have mineralcorticoid deficiency requiring replacement with fludrocortisone (0.1 mg/d.
Studies suggest that DHEA replacement in both women and men with primary and secondary adrenal insufficiency may improve small benefit in the quality of life, mood and sex drive.
Clinical practice guidelines recommend DHEA for replacement therapy in women with adrenal insufficiency who have low libido, fatigue, and symptoms of depression