 VIPomas are rare functioning neuroendocrine tumors that secrete vasoactive intestinal polypeptide (VIP).
VIPomas are rare functioning neuroendocrine tumors that secrete vasoactive intestinal polypeptide (VIP).
The VIPoma syndrome is caused by excessive, unregulated secretion of vasoactive intestinal polypeptide (VIP) by the tumor.
VIP is a 28 amino acid polypeptide that binds to high affinity receptors on intestinal epithelial cells, leading to activation of cellular adenylate cyclase and cAMP production.
It causes a net fluid and electrolyte secretion into the lumen, resulting in secretory diarrhea and hypokalemia.
VIPomas are detected in 1 in a million people per year.
VIPomas usually (about 90%) originate from the non-β islet cells of the pancreas.
They are sometimes associated with multiple endocrine neoplasia type 1.
VIPomas usually occur as isolated tumors, but in 5 percent of patients they are part of the multiple endocrine neoplasia syndrome type 1 (MEN1) and occur in association with parathyroid and pituitary tumors, gastrinoma, and other tumors.
Roughly 50–75% of VIPomas are malignant.
Even benign lesions are problematic because they tend to cause a specific syndrome: profound and chronic watery diarrhea and resultant dehydration, hypokalemia, achlorhydria, acidosis, flushing and hypotension hypercalcemia, and hyperglycemia.
This syndrome is the Verner–Morrison syndrome (VMS), WDHA syndrome (from watery diarrhea–hypokalemia–achlorhydria), or pancreatic cholera syndrome (PCS).
A watery diarrhea syndrome with elevated serum immunoreactive VIP has been described accompanying nonpancreatic tumors such as bronchogenic carcinomas, ganglioneuromas, pheochromocytomas, and a rare mastocytoma.
The major clinical features are prolonged watery diarrhea, with stool volume > 750 to 1000 mL/day.
Symptoms of hypokalemia and dehydration occur.
Half of the patients have relatively constant diarrhea while the rest have alternating periods of severe and moderate diarrhea.
One third have diarrhea < 1yr before diagnosis, but in 25%, diarrhea is present for 5 yr or more before diagnosis.
Lethargy, muscle weakness, nausea, vomiting and crampy abdominal pain are frequent symptoms.
Hypokalemia and impaired glucose tolerance occur in < 50% of patients.
Achlorhydria is also a feature.
Flushing similar to the carcinoid syndrome occur rarely.
VIPomas account for 2% to 7% of GEP neuroendocrine tumors and have an annual incidence of 1 per 10 million population.
The mean age of presentation is 49 years, with a slight female preponderance.
The majority of pancreatic VIPomas occur in the body and tail of the pancreas.
VIPomas are usually solitary with diameters of 1 to 7 cm, and 60-80% have metastasized at the time of presentation, typically to the liver and regional lymph nodes.
Approximately 60 to 80 percent of VIPomas have metastasized by the time of diagnosis [8,9].
Metastases to the kidneys, lungs, stomach, and mediastinum have also been reported.
The VIPoma syndrome reflect the physiologic effects of VIP and its related peptides.
Secretory diarrhea with large potassium, bicarbonate, and chloride losses, in conjunction with relaxation of the anal sphincter, can result in loss of large volumes of fluid: 3 L, but can be up to 20 L.
The diarrhea may be intermittent, thus delaying the diagnosis by an average of 3 years.
Vasodilator effects of VIP may result in hemodynamic collapse.
The hypokalemia results from intestinal losses and secondary hyperaldosteronism.
Other associated abnormalities: achlorhydria or hypochlorhydria, hypomagnesemia, hypercalcemia, glucose intolerance, and mild diabetes.
Approximately 20% of patients exhibit flushing of the head and trunk associated with a patchy erythematous rash.
The diagnosis of the VIPoma syndrome involves establishing the presence of secretory diarrhea, which is isotonic and persistent despite a 48- to 72-hour fast.
Tests include:
Blood chemistry tests (basic or comprehensive metabolic panel)
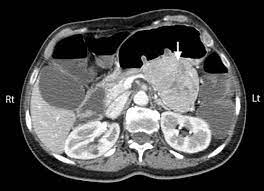
CT scan of the abdomen
MRI of the abdomen.
Stool examination for the cause of diarrhea and electrolyte levels
Vasoactive intestinal peptide (VIP) level in the blood.
A fasting plasma VIP concentration of greater than 200 pg/mL then confirms the diagnosis.
Several provocative tests, such as the pentagastrin test, have been recommended but not routinely used.
Besides the clinical picture, fasting VIP plasma level may confirm the diagnosis, and CT scan and somatostatin receptor scintigraphy are used to localize the tumor, which is usually metastatic at presentation.
ZOllinger-Ellison syndrome can be distinguished from the VIPoma syndrome by the presence of gastric acid hypersecretion, absence of metabolic acidosis, and minimal loss of potassium in the stool.
Ganglioneuroblastomas may secrete epinephrine and norepinephrine, resulting in raised urinary catecholamines.
VIPomas can be identified by ultrasound, CT, and radiolabeled somatostatin receptor and VIP scintigraphy.
Angiography and transhepatic venous sampling may be required if other methods have been unhelpful to establish a diagnosis.
The initial treatment depends on how severely the patient is affected and involves replacement of fluid and electrolytes.
The first goal of treatment is to correct dehydration.
Fluids are often given to replace fluids lost in diarrhea.
The next goal is to slow the diarrhea with medications that can help control diarrhea: Octreotide, blocks the action of VIP.
Somatostatin analogs inhibit the actions of VIP but also have independent inhibitory actions on intestinal secretion.
Tachyphylaxis to the actions of somatostatin analogs may occur.
Second line agents include: glucocorticoids, phenothiazines, prostaglandin synthesis inhibitors and lithium carbonate.
Surgical removal if possible, represents the best chance of a cure: one-third to one-half of patients, the tumor has spread by the time of diagnosis and cannot be cured.
Surgical debulking sometimes helps in symptom control.
Combination chemotherapy with 5-fluorouracil, temozolomide and streptozotocin has been shown to be effective.
Octreotide acetate controls diarrhea in up to 90% of patients with VIPomas.
Glucocorticoids reduce symptoms in 50%.
Systemic chemotherapy may be needed in cases of unresectable or progressive disease: Streptozocin, doxorubicin, fluorouracil, or a combination of these appears to be beneficial.
Local tumor resection is the treatment of choice.
In advanced disease, tumor debulking may relieve symptoms, but it is not effective in all cases.
Transarterial chemoembolization with chemotherapy-loaded materials may provide palliation in patients with extensive hepatic disease.
For metastatic disease, peptide receptor radionuclide therapy (PRRT) can be highly effective.
This treatment involves attaching a radionuclide (Lutetium-177 or Yttrium-90) to a somatostatin analogue (octreotate or octreotide).
The majority of VIPomas arise within the pancreas, and are classified as functioning pancreatic neuroendocrine islet cell tumors.
In adults, VIPomas are intrapancreatic in over 95 percent of cases.
Other VIP-secreting tumors have been reported, including lung cancer, colorectal cancer, ganglioneuroblastoma, pheochromocytoma, hepatoma, and adrenal tumors.
In children, VIPomas rarely arise in the pancreas.
VIP-secreting tumors in children typically occur in the sympathetic ganglia (ganglioneuroblastomas or ganglioneuromas) and the adrenal glands.
VIPomas are usually diagnosed between 30 and 50 years of age in adults and between two and four years of age in children.
Symptomatic pancreatic VIPomas are usually solitary, larger than than 3 cm in diameter, and arise in the tail of the pancreas in 75 percent of cases.
Other substances then VIP, such as prostaglandin E2, may occasionally be secreted by the tumors.
VIP activities include: vasodilation, inhibition of gastric acid secretion, bone resorption, and enhanced glycogenolysis and is responsible for flushing as well as laboratory findings of hypochlorhydria, hypercalcemia, and hyperglycemia in patients with VIPomas.
Most patients with VIPoma have VIPoma syndrome, which is also called the pancreatic cholera syndrome, Verner-Morrison syndrome, and the watery diarrhea, hypokalemia, and hypochlorhydria or achlorhydria (WDHA) syndrome.
VIPoma syndrome is characterized by watery diarrhea that persists with fasting.
Stools are tea-colored and odorless with stool volumes exceeding 700 mL/day. In 70 percent of patients.
Stool volume can exceed 3000 mL per day.
Abdominal pain is mild or absent.
Associated symptoms include flushing episodes in 20 percent of patients and symptoms related to hypokalemia and dehydration, such as lethargy, nausea, vomiting, muscle weakness, and muscle cramps.