 Autosomal dominant disorder associated with seizures, mental retardation, and skin lesions.
Autosomal dominant disorder associated with seizures, mental retardation, and skin lesions.
Prevalence approaches one in 6000 live births.
Tuberous sclerosis complex leads to activation of the mammalian target rapamycin complex 1.
A tumor suppressor syndrome caused by mutations in the tuberin gene (TSC2) or the hamartin gene (TSC1).
Mutations are found in more than 85% of patients.
Proteins encoded by the above genes form a tumor suppressor complex acting through the RAS homologue enriched in brain protein to limit activation of the mammalian target of rapamycin (mTOR) complex 1.
mTOR complex 1 is upregulated in this disease process leading to abnormal cellular growth, proliferation and protein synthesis.
Characterized by hamartomas in the brain, kidneys, lung, skin, and heart and retina.
TSC associated variety of tumor is in multiple organs including the brain, eyes, heart, lungs, kidneys, pancreas, thyroid colon due to mutations in either of the 2 genes TSC1 or TSC2.
80% of patients develop angiomyolipomas.
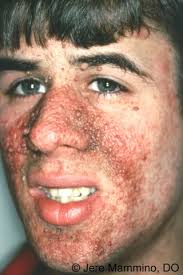
Associated with dermatologic findings of angiofibromas, connective tissue plaques and hypo pigmented lesions.
Lymphangioleiomyomatosis occurs sporadically in 5 women per million, and affects 30-40% of women with tuberous sclerosis complex.
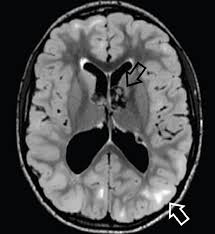
CNS hamartomas may lead to seizures.
Mental retardation and hypertension may occur
Angiomyolipomas are tumors rich in fat, muscle, and blood vessels that can lead to hemorrhage, infiltrates and renal failure.
Subependymal giant cell astrocytomas, slow-growing glioneuronal tumors that typically arise near the foramen of Monro developing 5-20% of patients.
Subependymal giant cell astrocytomas are associated with significant illness and death including acute hydrocephalus, the risk of which is directly related to the volume of the lesion.
Rapamycin complex one inhibitors are effective against tumors in patients with tuberous sclerosis complex including facial angiofibroma.
Everolimus, sirolimus available for Seizures in Tuberous Sclerosis