 Stiff-person syndrome (SPS)/stiff-man syndrome (SMS), is a rare neurologic disorder characterized by progressive rigidity and stiffness.
Stiff-person syndrome (SPS)/stiff-man syndrome (SMS), is a rare neurologic disorder characterized by progressive rigidity and stiffness.
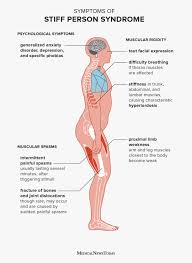
An adult onset acquired autoimmune disorder, associated with stiffness, gait dysfunction with falls, and an exaggerated startle response.
SPS primarily affects the truncal muscles and is superimposed by spasms, resulting in postural deformities.
It is associated with chronic pain, and impaired mobility.
Lumbar hyperlordosis are common symptoms.
SPS occurs in about one in a million people.
SPS is most commonly found in middle-aged people.
The age of onset varies from about 30 to 60, and it most frequently occurs in people in their 40s.
Greater than50% of persons with SPS have a history of other autoimmune conditions, with up to 30% having type one diabetes.
In a small number of patients it is related to a paraneoplastic condition (5-10%).
Stiffness and painful muscle spasms are the primary symptoms and begin in the paraspinal and abdominal muscles and progress to involve the proximal legs.
SPS variants primarily affecting a specific limb may occur.
Symptoms of SPS result from impaired gamma-amino butyric acid (GABA)-mediated inhibition of alpha motor neurons in the spinal cord and brain, leading to hyperactivity.
Sudden voluntary movement, physical touching, cold temperatures, emotional upset, and startle responses can provoke painful spasms, which may occur in clusters.
Falls occur due to superimposed stiffness and spasms with inability for patients to brace themselves.
SPS patients generally have glutamic acid decarboxylase (GAD) antibodies.
Glutamic acid decarboxylase (GAD) antibodies rarely occurs
Electromyography tests can help confirm the condition’s presence.
Benzodiazepines are the most common treatment for symptom relief from stiffness.
Other treatments include: baclofen, intravenous immunoglobin and rituximab.
There is encouraging therapeutic experience of haematopoietic stem cell transplantation for SPS.
Patients with stiff-person syndrome (SPS) experience progressive stiffness in their torso muscles.
Clinical exam does not show weakness with sensory loss and testing does not showing increase muscle tone.
70% of patients have increased deep tendon reflexes.
Examination may show an exaggerated startle response, abdominal and paraspinal muscles may appear hypertrophy and firm.
Paraspinal muscle rigidity may lead to lumbar hyperlordosis.
When truncated muscles become rigid and stiff the lumbar and abdominal muscles engage in constant contractions.
Initial stiffness occurs in the thoracolumbar paraspinal and abdominal muscles.
It later affects the proximal leg and abdominal wall muscles.
The stiffness leads to a change in posture, and the development of a rigid gait.
Persistent lumbar hyperlordosis often occurs as it progresses.
The muscle stiffness initially fluctuates, but eventually begins to consistently impair mobility.
Simultaneous contractions of agonist and antagonist muscle pairs can limit range of motion.
Patients sometimes become unable to walk or bend.
Chronic pain is common and worsens over time.
Occasionally acute pain occurs as well.
Symptoms are increased with exposure to stress, cold weather and infections.
Fewer symptoms are associated with increased sleep.
SPS patients experience spasms in the proximal limb and axial muscles.
There are contractions of agonist and antagonist muscles, and spasms usually last for minutes and can recur over hours.
SPS patients have extreme sensitivity to touch and sound.
Spasms are unpredictable and are often precipitated by fast movements, emotional distress, or sudden sounds or touches.
Occasional involvement of facial muscles, hands, feet, and the chest can be affected with unusual eye movements and vertigo.
There are brisk stretch reflexes and clonus findings.
Hypnagogic myoclonus can occur.
Tachycardia and hypertension are sometimes also present.
Spasms, may cause fearfulness, inability to work, leading to depression, anxiety, and phobias, including agoraphobia and dromophobia.
Most patients respond reasonably to their situations.
Paraneoplastic SPS tends to affect the neck and arms more than other variations,
progresses very quickly, is more painful, and is more likely to include distal pain than classic SPS.
Stiff-limb syndrome is a variant of SPS, developing into full SPS about 25% of the time.
Stiff-limb syndrome stiffness and spasms are usually limited to the legs and hyperlordoisis generally does not occur.
The stiffness begins in one limb and remains most prominent there.
Sphincter and brainstem issues often occur with stiff-limb syndrome.
Progressive encephalomyelitis with rigidity, is another variant of the condition, and includes symptoms of SPS with brainstem issues and autonomic disturbances.
Jerking SPS subtype begins like classical SPS and progresses with the development of myoclonus as well as seizures and ataxia in some cases.
SPS associated with high glutamic acid decarboxylase (GAD) antibody levels in blood.
About 80 percent of SPS patients have GAD antibodies, compared with about one percent of the general population.
However, the overwhelming majority of people who have GAD antibodies do not develop SPS.
This suggests the systemic synthesis of the antibody is not the sole cause of SPS.
Most SPS patients with high-titer GAD antibodies also have antibodies that inhibit GABA-receptor-associated protein.
Autoantibodies against amphiphysin and gephyrin are also sometimes found in SPS patients, which interact with antigens in the brain neurons and the spinal cord synapses, causing a functional blockade of the inhibitory neurotransmitter gamma-aminobutyric acid (GABA).
The GABA impairment probably causes the stiffness and spasms that characterize SPS.
There are low GABA levels in the motor cortexes of SPS patients.
The level of GAD antibody titers found in SPS patients does not correlate with disease severity.
In SPS patients, motor unit neurons fire involuntarily while at rest, particularly in the muscles which are stiff.
Motor neuron excess firing may be caused by malfunctions in spinal and supra-segmental inhibitory networks that utilize GABA.
Such involuntary actions show up as voluntary on EMG scans.
There are agonist and antagonist contractions.
Uncommonly patients with SPS and breast, ovarian, or lung cancer manifest paraneoplasticly as proximal muscle stiffness.
These cancers are associated with the synaptic proteins amphiphysin and gephyrin.
These patients tend not to have GAD antibodies.
The HLA class II locus makes patients susceptible to the condition.
Most SPS patients have the DQB1* 0201 allele, which is also associated with type 1 diabetes.
Diagnosis:
Underdiagnosis and misdiagnosis hinder epidemiological information about the condition.
SPS is diagnosed by evaluating clinical findings and excluding other conditions.
No specific laboratory test that confirms its presence.
Underdiagnosis and misdiagnosis are common.
The presence of antibodies against GAD is the best indication of the condition that can be detected by blood and cerebrospinal fluid (CSF) testing.
Anti-GAD is found in about 80 percent of SPS patients.
Anti-thyroid, anti-intrinsic factor, anti-nuclear, anti-RNP, and anti-gliadin are also often found in blood tests.
Electromyography (EMG) demonstrates involuntary motor unit firing in SPS patients.
EMG can confirm the spasms in distant muscles as a result of subnoxious stimulation of cutaneous or mixed nerves.
Responsiveness to diazepam helps confirm that the patient has SPS, as this drug will decrease stiffness and motor unit firing.
About 35 percent of SPS patients have type I diabetes.
Once SPS is diagnosed, poor response to conventional therapies and the presence of cancer indicate that it may be paraneoplastic process.
Similar symptoms to SPS occur with: myelopathies, dystonias, spinocerebellar degenerations, primary lateral sclerosis, neuromyotonia, some psychogenic disorders, tetanus, neuroleptic malignant syndrome, malignant hyperpyrexia, chronic spinal interneuronitis, serotonin syndrome, multiple sclerosis, Parkinson’s disease, and Isaacs syndrome.
Patients with SPS frequently have coexisting, anxiety, depression, agoraphobia, which may lead to misattribution of symptoms to psychiatric causes.
Diagnostic delay is common, it takes an average of six years after the onset of symptoms before the disease is diagnosed.
Clinical response to treatment with a benzodiazepine is a key diagnostic feature of stiff person syndrome.
Treatment:
The rarity of the disease complicates efforts to establish treatment guidelines.
Intravenous immune globulin has been found to be an effective therapy.
GABAA agonists, usually diazepam but sometimes other benzodiazepines, are the primary treatment.
Agents that increase GABA activity alleviate muscle stiffness caused by a lack of GABAergic tone.
Drugs that increase pathways that are dependent upon GABA and have muscle relaxant and anticonvulsant effects, often provide symptom relief.
As the condition worsens over time, patients generally require increased dosages, leading to more side effects.
Baclofen, a GABAB agonist, is generally used when individuals taking high doses of benzodiazepines have high side effects.
Intravenous immunoglobin is the best second-line treatment for SPS, decreasing stiffness and improves quality of life and startle reflex.
Steroids, rituximab, and plasma exchange have been used to suppress the immune system in SPS patients.
Plasma exchange benefits patients who do not respond to first-line therapy, and specifically beneficial for patients with anti-GAD65 positive SPS.
Hematopoietic stem cell transplantation has been performed in a few cases with severe anti-GAD positive SPS, resulting in clinical remission.
SPS is generally responsive to treatment, but the condition progresses and stabilizes periodically.
The quality of life declines as stiffness precludes many activities.
About 65 percent of patients are unable to function independently.
About ten percent of SPS patients require ICU at some point.
About 10% of SPS patients sudden death occurs, usually caused by metabolic acidosis or an autonomic crisis.