
 Spinal muscular atrophy (SMA) is a group of genetic diseases that damage and kill motor neurons, the nerve cells in the spinal cord and lower part of the brain that control voluntary muscle movement.
Spinal muscular atrophy (SMA) is a group of genetic diseases that damage and kill motor neurons, the nerve cells in the spinal cord and lower part of the brain that control voluntary muscle movement.
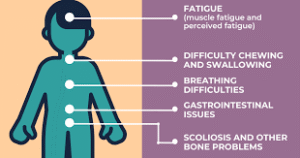
As these motor neurons are lost, muscles weaken and waste away over time, which can affect movement, swallowing, and breathing.
Most types of SMA are caused by a mutation or deletion in both copies of the survival motor neuron 1 (SMN1) gene on chromosome 5.
A deficiency of the SMN protein, which is necessary for motor neuron health and function.
The severity of SMA is largely determined by the number of copies of a neighboring gene, SMN2.
SMN2produces a small amount (about 10%) of functional SMN protein.
More SMN2 copies generally lead to milder symptoms.
SMA is typically inherited in an autosomal recessive pattern.
Carriers usually have no symptoms.
SMA is classified into types based on the age of onset and the maximum motor function achieved.
Type 0 (Prenatal): Symptoms begin before birth, with decreased fetal movement.
Infants have severe weakness and respiratory failure, typically surviving only a few weeks to months.
Type 1 (Severe Infantile Onset): The most common and severe form, with onset from birth to 6 months.
Infants have profound hypotonia (floppy muscles), poor head control, and difficulty feeding and breathing.
Without treatment, most children do not survive past age two.
Type 2 (Intermediate): Symptoms appear between 6 and 18 months.
Children can sit without support but cannot stand or walk independently.
They often develop scoliosis and may have respiratory issues.
Life expectancy is often into young adulthood.
Type 3 (Mild Juvenile Onset): Symptoms appear after 18 months or in early childhood.
Children can walk independently but may have difficulty running, climbing stairs, or getting up from the floor. Most maintain a normal life expectancy but may require a wheelchair later in life.
Type 4 (Adult Onset): Rare and mild, with symptoms usually beginning in adulthood (after age 21 or 30).
Symptoms include mild to moderate leg muscle weakness and tremors.
Life expectancy is normal.
Diagnosis and Treatment
Diagnosis typically involves a physical exam, medical history, family history and genetic testing to check for SMN1 gene mutations.
In the U.S., most newborns are now screened for SMA, allowing for early diagnosis and treatment before symptoms appear.
There is no cure for SMA
Standard supportive care includes:
Physical and occupational therapy to manage muscle weakness and prevent joint contractures and scoliosis.
Nutritional support, which may involve a feeding tube if swallowing difficulties arise.
Breathing support, such as non-invasive ventilation or assisted coughing devices.
Assistive devices like braces, walkers, and wheelchairs to aid independence.
Disease-modifying therapies have been approved by the FDA that increase the amount of functional SMN protein: Nusinersen (Spinraza®): An injectable medication administered into the spinal fluid that modifies SMN2 gene splicing to produce more functional SMN protein.
Onasemnogene abeparvovec (Zolgensma) A one-time gene replacement therapy for children under 2 years of age that delivers a functional copy of the SMN1 gene.
Risdiplam (Evrysdi) An oral liquid medication that also increases SMN protein production from the SMN2 gene.
Early treatment, especially before symptom onset, has shown significant improvements in motor function and survival rates.