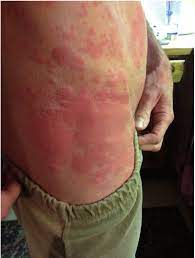
Schnitzler syndrome or Schnitzler’s syndrome (SS) is a rare disease characterized by onset around middle age of chronic hives and periodic fever, bone pain and joint pain, weight loss, malaise, fatigue, swollen lymph glands and enlarged spleen and liver.
It is considered a late onset autoinflammatory disorder and is generally treated with anakinra, which inhibits interleukin 1.
This treatment controls the condition but does not cure it.
SS does not shorten survival.
There is a slightly higher incidence among men than women.
The duration of urticarial skin rashes varies from less than 24 to 48 hours.
Wheals are typically the presenting symptom, and other common symptoms include fever, arthralgias, and bone pain.
Comprises up to 1% of patients with chronic urticaria or mono IGM gammopathy.
Median age of onset 51 years with a male to female ratio of 1.5.
It is associated with chronic recurrent wheels that do not resolve with antihistamines, along with fever, arthralgia, elevated CRP, and characteristic neutrophilic infiltrates on skin biopsy.
Associated angioedema is rare.
Monoclonal IgM gammopathy is seen in 85% of patients and IgG gammopathy in 7% of patients.
Skin biopsy reveals a neutrophilic dermal infiltrate.
About 15% of people develop complications.
The typical age of onset is at around 55 years.
Approximately 55 to 75% of patients have bone or joint pain.
The femur, tibia, and iliac bones are most commonly infected.
Bone scan they show bone remodeling.
Symptoms are recurrent hives, mostly on the torso and limbs, often with recurring fever, joint pain, bone pain, muscle pain, headache, fatigue, and loss of weight.
Fever is common and typically intermittent.
Fatigue is common during fever episodes.
Lymphadenopathy, weight, loss, hepatomegaly, or splenomegaly may be present.
Patients may have neutrophilic leukocytosis, thrombocytosis, elevated bone remodeling markers, such as alkaline phosphatase and osteocalcin.
Patients are at increased risk for a lymphoproliferative disorder that develops in approximately 15 to 20% of patients, typically 10 years or more after the initial disease manifestation: lymphoplasmacytic lymphoma is the most common of these disorders.
Evidence for an autoinflammation caused by an overactive innate immune system that is mediated by excessive production of interleukin-1, interleukin-6, and tissue necrosis factor alpha, coupled with a reduction in T cell function that can be restored by interleukin-1 blocking therapies.
A somatic mutation in MYD 88 L265P is reported in 30% of patients with Schnitzler syndrome.
Interleukin-1 receptor antagonist, anakinra may result in complete remission in more than 80% of patients.
Patients with SS have a high concentration of specific gamma-globulins-monoclonal gammopathy of the IgM type.
It almost always has light chains of the κ-type.
A variant in IgG appears to be one-tenth as common.
Signs of inflammation are often present, with an increased white blood cell count and a raised erythrocyte sedimentation rate , C-reactive protein and elevated serum amyloid protein.
Patients may have anemia of chronic disease.
Bone abnormalities can include increased density or osteosclerosis.
Dagnostic criteria, the Lipsker criteria: hives, the presence of monoclonal IgM, and at least 2 of the following: fever, joint pain or arthritis, bone pain, swollen lymph nodes, enlarged spleen or liver, elevated erythrocyte sedimentation rate, high levels of white blood cells, and findings of problems in bone imaging.
Strasbourg criteria: hives and the presence of monoclonal IgM or IgG, and two of the following, recurrent fevers, abnormalities in bone imaging, with or without bone pain, findings of neutrophil infiltration in a skin biopsy, high levels of white blood cells or C-reactive protein.
Differential diagnosis: autoimmune or autoinflammatory disorders such as adult-onset Still’s disease, angioedema, hematological disorders such as lymphoma or monoclonal gammopathy of undetermined significance (MGUS), other causes of hives, cryoglobulinemia, mastocytosis, chronic neonatal onset multisystem inflammatory disease or Muckle–Wells syndrome.
Treatment:
ANtihistamines, nonsteroidal, anti-inflammatory drugs, colchicine, at oral glucose typically have limited efficacy in Schnitzler syndrome.
Drugs that inhibit interleukin 1 activity have been the preferred treatment: anakinra, canakinumab , immunosuppressant drugs such as corticosteroids, cyclooxygenase inhibitors, and interferon alpha
Anakinra is a first line treatment in patients with a documented diagnosis of Schnitzler syndrome.
IL-1/receptor antagonists are effective in most patients.
Anakinra is highly and rapidly effective for inducing complete remission of Schnitzler syndrome.
Anakinra is not curative, however; symptoms recur soon after treatment stops.
In around 15-20% of people, a lymphoproliferative disorder as a complication, most commonly Waldenström’s macroglobulinemia, develops.
AA amyloidosis has also been reported in people with Schnitzler syndrome.
The life span has not been shown to differ much from the general population.
Other agents reported to have advocacy include tocilizumab, and rilonacept.