 Characterized by mental retardation, short stature, hypotonia, hypogonadism, small hands and feet and obesity.
Characterized by mental retardation, short stature, hypotonia, hypogonadism, small hands and feet and obesity.
The main characteristics of PWS include extreme, insatiable appetite, mild to moderate developmental delay, hypogonadism resulting in delayed to no puberty, and hypotonia.
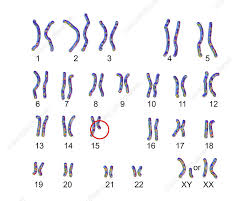
Prader-Willi syndrome is caused by the loss of active genes in a specific part of chromosome 15, the q11-q13 region.
It is the prototypical example of a genetic disorder that can cause insatiable hunger in the development of severe obesity during childhood.
People normally have two copies of this chromosome in each cell, one copy from each parent.
Prader–Willi syndrome occurs when the paternal copy is partly or entirely missing.
65-70% of cases have a deletion of q12 in the long arm of chromosome 15, del(15)(q11.2q13).
Deletion affects paternally derived chromosome.
A rare genetic disorder in which seven genes on chromosome 15 (q 11–13) are deleted or unexpressed on the paternal chromosome.
The genes in this region are normally active on the paternal copy of the chromosome and are inactive on the maternal copy.
Therefore, a person with a deletion in the paternal chromosome 15 will have no active genes in this region.
In about 25% of cases, a person with Prader–Willi syndrome has two maternal copies of chromosome 15 in each cell instead of one copy from each parent.
This phenomenon is called maternal uniparental disomy.
Because some genes are normally active only on the paternal copy of this chromosome, a person with two maternal copies of chromosome 15 will have no active copies of these genes.
In a small percentage of cases the condition is caused by an abnormality in the DNA region that controls the activity of genes on the paternal chromosome 15.
Because patients almost always have difficulty reproducing, Prader–Willi syndrome is generally not hereditary.
Prader–Willi syndrome is specifically linked to an epigenetic pattern in which the paternal copy in the chromosomal region is erroneously deleted, and the maternal loci is inactivated by over methylation.
Characteristic of PWS is low muscle tone, short stature, incomplete sexual development, cognitive disabilities, behavior problems, and a chronic feeling of hunger that can lead to excessive eating and life-threatening obesity.
Children with Prader-Willi syndrome have complications of severe obesity, including type two diabetes, heart failure, and they rarely survive beyond 25 to 30 years of age.
The incidence of PWS is between 1 in 25,000 and 1 in 10,000 live births.
The paternal origin of the genetic material affected in the syndrome of the particular region of chromosome 15 involved is subject to parent-of-origin imprinting.
For a number of genes in this region, only one copy of the gene is expressed while the other is silenced through imprinting.
In PWS the maternal copy of genes is usually imprinted and thus is silenced, while the mutated paternal copy is not functional.
Most people have one working and one silenced set of these genes, people with PWS have a non-working set and a silenced set.
If the maternally derived genetic material from the same region is affected instead, the sister Angelman Syndrome occurs.
With early diagnosis and interventions, the obesity rate among children has decreased to be similar to the typical population.
With behavioural therapy and other treatments, the effects of the syndrome can be reduced.
The symptoms can range from poor muscle tone during infancy to behavioral problems in early childhood.
Some findings found in infants include: poor muscle tone, lack of eye coordination, almond-shaped eyes; infants may not have a strong sucking reflex, cry is weak, difficulty waking up, and a thin upper lip.
Other aspects include hypotonia and abnormal neurologic function, hypogonadism, developmental and cognitive delays, hyperphagia and obesity, short stature, and behavioral and psychiatric disturbances.
A down-turned mouth can be part of the presentation of Prader-Willi syndrome.
Associated with reduced fetal movement, abnormal fetal position, occasional polyhydramnios, often breech presentation or caesarean births.
Associated with learning disabilities.
Patients are prone to diabetes mellitus.
Associated with extreme flexibility
Associated with prominent nasal bridge.
Associated with small hands and feet with tapering of fingers.
Associated with soft skin, which is easily bruised.
Associated with excess fat, especially in the central portion of the body.
Prader-Willi syndrome is associated with decreased response to satiety that may promote development of obesity.
High, narrow forehead
Thin upper lip
Downturned mouth
Almond-shaped eyes
Light skin and hair relative to other family members
Lack of complete sexual development
Frequent skin picking
Striae
Delayed motor development
At risk of learning and attention difficulties.
5%: IQ above 85 (high to low average intelligence)
27%: IQ 70–85 (borderline intellectual functioning)
39%: IQ 50–70 (mild intellectual disability)
27%: IQ 35–50 (moderate intellectual disability)
1%: IQ 20–35 (severe intellectual disability)
<1%: IQ <20 (profound intellectual disability)
Most individuals (50–65%) fall within the mild/borderline/low average intelligence range.
Some children have IQs >110 and function normally in school.
Children are often strong in visual organization and perception, including reading and vocabulary.
Affected children’s spoken language is sometimes affected by hypernasality, is generally poorer than their comprehension.
Auditory information processing, sequential processing, arithmetic and writing skills, visual and auditory short-term memory and auditory attention span are poor, and such deficits remain throughout adulthood.
Frequently associated with insatiable appetite, often resulting in morbid obesity.
Prader-Willi syndrome has a decreased response to satiety that may promote development of obesity.
Prader-Willi syndrome is the most common genetic cause of morbid obesity in children.
Genetic abnormalities in chromosome 15 disrupt the normal functioning of the hypothalamus and may be related.
In patients with Prader-Willi syndrome
the hypothalamus nerve cells produce oxytocin, a hormone thought to contribute to satiety, have been found to be abnormal.
Patients have high ghrelin levels, which are thought to directly contribute to the hyperphagia, and obesity.
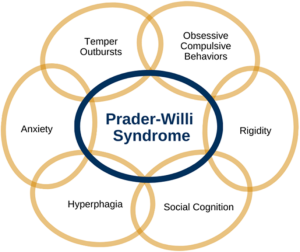
Patients with PWS experience compulsive behavior and anxiety.
5-10% of patients experience hallucinations, paranoia and depression.
Behavioral problems develop in early childhood in 70–90% of cases.
Behavioral problems include:
stubbornness, temper tantrums, controlling and manipulative behavior, difficulty with change in routine, and compulsive-like behaviors.
Patients have short stature, are obese with abnormal body composition, have reduced fat free mass, have reduced lean body mass (LBM) and total energy expenditure, have decreased bone density, and hypogonadism.
In males undescended testes may occur.
In females benign premature adrenarche may occur.
Commonly associated with development of strabismus.
Caused by the deletion of the paternal copies of the SNRPN and necdin genes along with clusters of snoRNAs.
These are on chromosome 15 located in the region 15q11-13, and this region may be lost by several genetic mechanisms which, in the majority of instances occurs through chance mutation.
The maternally inherited copies of these genes are virtually silent, only the paternal copies of the genes are expressed.
PWS results from the loss of paternal copies of this region.
The risk to siblings is <1% if the affected child has a gene deletion or uniparental disomy, up to 50% if the affected child has a mutation of the imprinting control region, and up to 25% if a parental chromosomal translocation is present.
Prenatal testing is possible for the genetic mechanisms.
Deletion of the 29 copies of the C/D box snoRNA SNORD116 (HBII-85) has been shown to be the primary cause of Prader–Willi syndrome.
PWS affects approximately 1 in 10,000 to 1 in 25,000 newborns.
There are more than 400,000 people who live with PWS around the world.
It is traditionally characterized by hypotonia, short stature, hyperphagia, obesity, behavioral issues, small hands and feet, hypogonadism, and mild intellectual disability.
With early diagnosis and treatment the prognosis is beginning to change.
It is a spectrum disorder and symptoms can range from mild to severe and may change throughout the person’s lifetime.
The syndrome is diagnosed through genetic testing.
Genetic testing is recommended for newborns with pronounced hypotonia.
Early diagnosis and early intervention as well as the early use of growth hormone supports linear growth and increased muscle mass, and may lessen food preoccupation and weight gain.
Daily recombinant growth hormone injections are indicated.
In Prader–Willi syndrome GH can help children with height, weight, body mass, strength, and agility.
With GH increase of growth rate, especially in the first year of treatment, and improved body composition, higher muscle mass, lower fat mass, improved weight management; increased energy and physical activity; improved strength, agility, and endurance; and improved respiratory function have been reported.
Diagnosis is by genetic testing, specifically DNA-based methylation testing to detect the absence of the paternally contributed Prader–Willi syndrome/Angelman syndrome (PWS/AS) region on chromosome 15q11-q13.
Genetic testing detects over 97% of cases.
Often misdiagnosed as other syndromes.
Has no cure.
Patients require therapy to improve muscle tone, speech and occupational therapy as indicated.
Children benefit from a highly structured learning environment.
The largest problem associated with the syndrome is severe obesity.
Physical activity is encouraged to optimize strength and promote a healthy lifestyle.
Daily recombinant growth hormone injections are indicated for children, as it supports linear growth and increased muscle mass, and may lessen food preoccupation and weight gain.
Obstructive sleep apnea is a common complication of obesity.
Unsuccessful treatment for the obesity is gastric bypass.
Patients have a very high tolerance to pain.
Behavior and psychiatric problems are treated with parental education and training.
Serotonin agonists are effective in lessening temper tantrums and improving compulsivity.