 A rare autosomal recessive progressive neuromuscular disease.
A rare autosomal recessive progressive neuromuscular disease.
A metabolic myopathy caused by a deficiency of acid alpha glucosidase, an enzyme that degrades lysosomal glycogen.
It is caused by a genetic deficiency of acid alpha-glucosidase (GAA) a lysosomal enzyme that converts glycogen into glucose.
More than 600 pathogenic variants in GAA have been reported.
A glycogen storage disorder characterized by multiple organ involvement.
It results in glycogen accumulation, primarily in skeletal, cardiac, and other muscles.
There are two types of Pompei disease: infantile onset associated with near complete deficiency of acid alpha glucosidase enzyme activity with clinical onset before 12 months of age and severe cardiac involvement and; late onset and Pompei disease caused by partial enzyme deficiency of less than 40% normal activity, with the age of onset, ranging from infant to late adulthood.
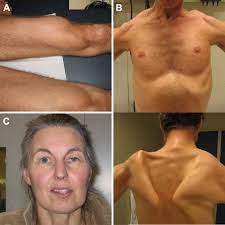
Clinical presentations of the process are diverse, and often include proximal muscle weakness, and respiratory insufficiency, with cardiac involvement on rare occasions.
Patients with late onset Pompei disease typically present with progressive myopathy and respiratory symptoms.
Other manifestations include vascular malformations, oral, gastrointestinal abnormalities of dysphasia, GERD. constipation, dysmotility, cardiovascular abnormalities of a arrhythmia, conduction abnormalities, cardiomyopathy, cognitive and emotional disturbance.
Glycogen may accumulate in neural tissues, and a pair of phrenic nerve function, as well as skittle to muscle strength.
It is almost invariably associated with severe and progressive, physical disability and premature death, particularly among patients with infantile onset disease.
Characterized by progressive muscle weakness and loss of respiratory function, leading to early death.
If left untreated late onset Pompei disease leads to early death from progressive decline in pulmonary function and muscle strength, inversely related to acid alpha glucosidase enzyme activity.
In the classic infantile form glycogen is deposited in the heart, skeletal and respiratory muscles causing severe cardiomyopathy, hypotonia, and respiratory failure, leading to death within the first year of life.
In children and adults the disease is variability in disease progression.
Without treatment patients rarely severe survive beyond one year of age.
Most deaths due to respiratory failure.
Treatment with enzyme replacement therapy with glucosidase alfa.
Infants treated with alglucosidase alfa have improvement in survival and motor outcomes and use in children 8 years or older was associated with improved walking and stabilization in pulmonary function over 18 months (van der Ploeg AT).
Alphaglucosidase approved based on the results of a randomized phase 3 trial manifesting improved six minute walking tests.
Alphaglucosidose is the standard treatment leading to prolonged survival among patients with infantile onset Pompe’s disease, although treatment does not target nervous system involvement.
Enzyme replacement prolongs survival, reduces cardiomyopathy, improves motor and respiratory function, but has severe limitations and drawbacks.
The recombinant human GAA is highly immunogenic and elicits immune responses.
The drugs uptake and efficacy in some affected tissues are poor, resulting in emergence of residual phenotypes and secondary abnormalities.
After an initial improvement of symptoms, disease progression may resume or worsen in some patients.
The need for infusions every two weeks negatively affects patient’s quality of life.
This enzyme cannot cross the blood brain barrier and is unlikely to affect levels of glycogen in the brain.
Recombinant enzyme,idase alpha plus miglustat is an alternative therapy with greater pulmonary function than alglucossidase alpha therapy.
AAV9 – mediated gene therapy with adenoassociated virus has demonstrated gene therapy efficacy in this disease.