 Chronic process of the skeleton with one or more areas of aggressive osteoclast mediated bone resorption preceding imperfect osteoblast mediated bone repair.
Chronic process of the skeleton with one or more areas of aggressive osteoclast mediated bone resorption preceding imperfect osteoblast mediated bone repair.
Associated with accelerated bone turnover.
A focal disorder of bone metabolism involving increased skeletal resorption, formation and mineralization.
Can occur in 1 or several bones, monostotic or polyostotic disease.
Pelvis, femur, lumbar spine, and skull are the most common involved sites.
Associated with disorganized collagen assembly causing bone enlargement and deformity.
And unknown trigger speeds up bone resorption by osteoclasts.
Bone formation by osteoblasts is also increased, and the bone produced is abnormal, larger, weaker, and prone to fracture.
Begins as a area of destructive osteoclasts at one end of a bone that slowly advances to disrupt the entire structure.
Deranged skeletal remodeling causes one expansion, softening associated with pain and sometimes fracture, deformity and rarely malignant transformation.
Osteoblasts or osteoclasts may predominate at any given time, resulting in sclerosis, or lytic lesions.
Lytic lesions may become sclerotic later in the process.
It is common for sclerotic and lytic changes to exist within the same bone.
Unaffected bone completely within the normal range.
Etiologies include genetics, environmental influences, toxins, zoonotic infections, viruses, low calcium and vitamin D intake in childhood.
Unknown cause with 10% positive family history.
Results as a loss of the usual tight control of bone cell function with veractivity of bone forming cells and bone resorbing cells.
Osteoblast hyperactivity leads to the laying down which is disorganized in structure, prone to deformity and mechanically inadequate.
Increased osteoblast activity can lead to bone expansion, premature arthritis, if it affects the articular surface, nerve compression, and bone pain.
Second most common bone disorder behind osteoporosis.
Prevalence of 5% of population older than 55 years.
Rare before age of 55, and increases in prevalence thereafter, affectingly about 5% of women and 8% of men by the 8th decade of life.
Most common in the United Kingdom, and rare in Africans, people from the Indian subcontinent and Asians.
Susceptibility probably related to a genetic basis.
Manifests in middle or advanced age with a slight male predominance.
Approximately 1% of population over the age of 40 years affected in the U.S.
Juvenile form rare autosomal recessive process with deafness, fractures, bone deformities, accelerated bone turnover and due to osteoprotegerin deficiency.
Incidence increases with age.
Osteoclast numbers are increased with increase in bone resorption and formation of cavities which are filled by osteoblastic activity resulting in structurally weakened bone.
Increased bone remodeling results in osteopenia and osteosclerosis.
Bisphosphonates most widely used treatment.
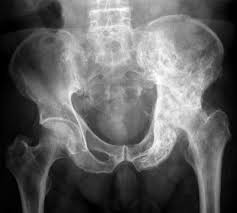
Most patients have multiple site bone involvement with pelvis, lumbar spine, and femur involved in 75% of cases.
Preferentially involves the axial skeleton, most frequently the pelvis with 70% of cases, femur 55% of cases, lumbar spine 53%, skull 42%, and tibia 32% (Siris ES et al).
More than 95% of patients are without symptoms.
Most common clinical problems include pain, skeletal deformities, and fractures.
30% of patients have symptoms for more than 10 years before diagnosis.
Disease progression occurs at about 1 cm per year along a long bone, indicating that most patients have active disease for a decade or more clinical presentation.
Skeletal deformity with bone enlargement can be seen in limbs, skull and facial bones.
Generally the disease progresses, until the entire bone is involved.
The number of bones involved remains steady throughout the illness, and the process does not spread from bone to bone.
Fractures of the femur, tibia and forearm are the most common sites of fractures and account for 90% of long bone fractures.
Patients with more than 30% of bone involvement can have increased cardiac output leading to heart failure.
Can rarely lead to bone sarcoma and accounts for most cases of bone sarcoma in patients over the age of 50 years.
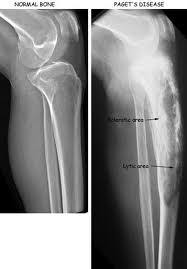
Radiographic changes include osteolytic, sclerotic changes with thickened cortices of long bones and accentuation of trabecular markings.
The pelvis is involved in two thirds of patients making it the most common site of bone involvement.
Serum alkaline phosphatase most often used to measure the extent of skeletal involvement.
Bone scan highlights areas of affected bone and can help define the extent of disease.
Radiographs may show cortical thickening, bowing, joint narrowing, and classic blade of grass sign may be seen in long bones.
Isolated increase in the alkaline phosphatase in a healthy person suggests the diagnosis.
Decreases bone resorption and formation with effects lasting for many months or years following cessation of therapy.
Suspected that the disease process has infection as a potential trigger based on the fact that there are intra-nuclear inclusion bodies resembling paramyxovirus nucleocapsid in Paget associated osteoclasts (Rebel A et al)..
Invariable responds to medical management.
Treatment is typically aimed at relieving symptoms and preventing complications, such as bone pain, fractures, and deformities.
Long-term treatment results in resumption of lamellar bone formation and a more normal radiographic appearance of bone with biochemical control and clinical improvement.
Paget’s disease associated with osteosarcoma more frequent among males, 1.58:1.
The incidence of malignant transformation of Paget’s disease is approximately 1%.
Osteosarcomas arising from Paget’s disease have a poor prognosis, with no recent improvements in treatment or survival.
Both Paget’s disease and osteosarcoma demonstrate a loss in heterozygosity, involving to varying degrees, of the distal end of chromosome 18 (McNairm JD et al).
Treatment of choice is nitrogen-containing bisphosphonates.
Risedronate 30 mg daily for two months or alendronate 40 mg daily for six months are effective therapies.
Zoledronic acid 5 mg intravenously is effective.
The mainstays of treatment include: Bisphosphonates:These are the first-line treatment for Paget’s disease.
They help in slowing down bone turnover.
Commonly used bisphosphonates include: – Alendronate – Risedronate – Zoledronic acid (often given as a single intravenous infusion) – Pamidronate
Analgesics or nonsteroidal anti-inflammatory drugs (NSAIDs) may be prescribed to manage bone pain.
Calcitonin hormone can be used if bisphosphonates are not suitable, as it helps regulate bone metabolism.
In cases where there are complications such as severe arthritis, fractures, or deformities, surgical intervention may be necessary.
Regular follow-up with blood tests to monitor alkaline phosphatase levels, a marker of bone turnover, may be required to assess the activity of the disease and response to treatment.
Encouraging a healthy lifestyle, including adequate intake of calcium and vitamin D, regular exercise, and fall prevention strategies, can be beneficial.
Physical Therapy can help maintain mobility and strengthen muscles around affected bones.