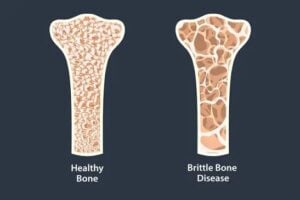
 Brittle bone disease autosomal dominant genetic disorder associated with bone fragility and fracture susceptibility.
Brittle bone disease autosomal dominant genetic disorder associated with bone fragility and fracture susceptibility.
The most prevalent heritable disorder of connective tissue.
Hallmark features are bone fragility, blue sclera, and dental disease.
Type I-XII with range of symptoms the mild to a perinatal lethal process.
Types I-V are inherited in an autosomal dominant manner.
Types VI-XII are autosomal recessive.
Seven recessive forms are caused by defect in genes whose protein products interact with collagen for folding or post translational modification.
To rare defects affect bone mineralization, but also decrease collagen production.
Approximately 60% of individuals with mild disease have de novo mutations.
A rare disorder occurring in 1 in 15-20,000 births.
A generalized connective tissue disorder with major manifestation in bone with skeletal fragility growth deficiency.
Mutations in the major structural protein of bone and skin, type I collagen, causes most cases.
Heterozygous mutations affecting the primary collagen structure causes moderate to severe osteogenesis imperfecta.
Approximately 90% of patients with types I, II, III, and IV have and identifiable mutation in COL1A1 or COL1A2.
Missense mutation in COL1A1 Gene along with COL1A2 encodes type I collagen.
A genetic disorder caused by mutations either in the genes encoding one of the two types of alpha chains of type I collagen, making up 90% of cases of classic osteogenesis imperfecta, or in genes with protein product’s involved in the proper folding in processing of type I collagen, making up 10% of cases.
No patients with types V, VI, and VII, have an identifiable mutation in COL1A1 or COL1A2.
COL1A1 and COL1A2 code for the alpha-1 and alpha 2 chains of type one collagen, the most abundant protein of bone, skin and extracellular matrices.
85-90% of cases are caused by structural or quantitative mutations in the collagen genes,
A genetically and phenotypically heterogeneous process with variable modes of inheritance and penetrates.
Associated with many mutations in type I collagen genes and other genes involved in bone integrity.
Decreased collagen production causes mild osteogenesis imperfecta.
Genetic changes reducing production of normal collagen leads to bone fragility, thinning of the sclera causing visualization of underlying bluish choroid.
Blue sclera is not pathognomonic for osteogenesis imperfecta, and differential diagnosis includes alkaptonuric ochronosis, and Ehlers-Danlos syndrome among others.
Should be considered in young patients with multiple fractures and osteopenia.
Classic Type IA disease is an autosomal dominant disorder associated with one of many mutations of the type I collagen gene.
Recessive form suggested to occur in families with normal parents with more than one child with severe bone dysplasia.
Patients may present in early infancy or later in life with single or multiple fractures.
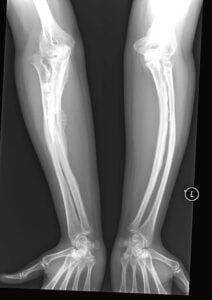
Diagnosis largely based on clinical and x-ray findings.
Mild OI may be difficult to discriminate from early onset osteoporosis in adults or physical abuse in children.
Fractures from mild trauma, bowing deformities in long bones and growth deficiency are hallmark features.
Skeletal features can include macrocephaly, flat face and triangular faces, dentigenesis imperfecta, chest wall deformities, such as pectis excavatum for carinatum, barrel chest, scoliosis o kyphosis.
Bone x-rays reveal generalized osteopenia, some combination of long bonebowing, under tubulation and flaring, narrow thoracic apex and vertebral compression.
OI caused by a collagen defect types I-IV, bone histomorphometry reveals low bone volume and trabecula number with high turnover kinetics.
Histological changes are distinctive in the diagnosis of type V and VI.
A generalized connective tissue disorder with findings to nonskeletal findings include:blue sclera, hearing loss, decreased pulmonary function, and cardiac valvular regurgitation.
Laboratory evaluation includes measurements of serum calcium, outgoing phosphatase, 25-hydroxyvitamin D, phosphorus, and parathyroid hormone levels, and skeletal bone survey.
Bone biopsy is generally not required for diagnosis but maybe an adjunct to the diagnosis of OI types V and VI.
Genetic counseling of family members is required, and prenatal testing for patients at risk pregnancies should be considered.
Bisphosphonate therapy to prevent and treat fractures is a supportive therapy, but have not not convincingly been shown to reduce the risk of fracture.
In a randomized trial of adults with OI found that teriparatide plus zoledronic acid did not reduce fracture risk compared to standard care, despite significant increasing bone mineral density, suggesting the importance of reduced bone quality rather than low bone density in the pathogenesis of fracture.