Characterized by blood vessel angiomatous lesions appearing as blood blisters on mucous membranes, lips and gastrointestinal tract.
Affects approximately one in 3800 persons and is the second most common inherited bleeding disorder.
Four criteria for definitive diagnosis: epistaxis, mucocutaneous telangiectasis, visceral arteriovenous malformations, and a positive family history.
Vascular lesions typically involved skin, mucous membranes, and visceral organs including the lung, liver, GI tract, brain, spinal cord, and other organs.
Arteriovenous malformations in the nasal cavity and GI tract have a propensity for spontaneous and recurrent bleeding, resulting in chronic recurrent epistaxis and Gi bleeding.
Bleeding from lesions increases in frequency with age and may result in chronic blood loss and iron deficiency.
Dominant inherited genetic vascular disorder characterized by recurrent epistaxis, cutaneous telangiectasia, and visceral arteriovenous malformations.
Autosomal dominant disorder affecting one in 5000 individuals.
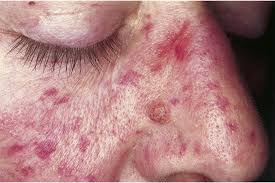
Nearly all patients developed telangiectasia involving the face, oral cavity, and hands.
Bleeding is the most common presenting symptom of telangiectasia.
Iron deficiency develops in most patients.
HHT bleeding usually increases over the lifespan of affected patients and results in need of frequent iron infusions, red blood, cell transfusions, or both.
Epistaxis affects approximately 50% of patients by age 20 in virtually all by age 50.
Severity of epistaxis vary widely among patients and among close related family members.
GI bleeding is a result of diffuse telangiectasias located along the length of the GI tract.
Epistaxis and GI bleeding can lead to severe blood loss, anemia, and requirement for red blood cell transfusions.
Epistaxis and GI bleeding person with age.
81% have GI telangiectasia.
Process may involve the lungs, gastrointestinal tract, liver and brain.
Criteria for diagnosis are recurrent epistaxis, telangiectasias, family history, and visceral lesions.
As many as 10-15% of patients have severe bleeding and anemia refractory to conventional therapy.
Patients with HHT may have a shorter overall survival than the general population.
Patients can have widespread telangiectasia , usually evident on the face, neck, upper trunk, and arms and legs or randomly distributed, and the positive family history, but mucosal lesions, epistaxes, and visceral arteriovenous malformations are absent.
Patients with arteriovenous malformations typically have symptoms related to blood shunting.
Up to 74% of patients have hepatic involvement, but only 8% or so, mainly women, have symptomatic liver shunting.
Liver vascular malformations can result in high output cardiac failure, portal hypertension, and biliary necrosis.
Liver arteriovenous malformations are present it up to 70% of patients.
Shunts can develop between hepatic artery and hepatic veins, the hepatic artery and portal vein, or the portal vein and hepatic veins.
Fewer than 10% of liver AV malformation patients become symptomatic.
Patients may present with portal hypertension, encephalopathy, or high output cardiac failure.
Associated with 2 genes: ENG, endoglogin, and ACRLV1, activin receptor like kinase 1,ALK-1.
Mutations in the one or both of the above genes account for most cases.
MADH4 mutations are responsible for juvenile polyposis/HHT overlap syndrome.
ENG, and ACVRL1 encode endothelial cell transmembrane proteins components of the receptor complexes for growth factors of the transforming growth factor beta superfamily (TGF-beta)..
Diagnosis based on clinical findings known as the Curaçao criteria: spontaneous recurrent nosebleeds, telangiectasia of the lips, oral cavity, fingertips, or nose: arteriovenous malformations of the gastrointestinal tract, lungs, brain, liver or spine: and a history of HHT in a 1st degree relative.
If three of the criteria are present the patient has a definitive diagnosis, and if two criteria are present they have suspected disease.
Bevacizumab is an effective treatment for patients with severe anemia related to epistaxis and gastrointestinal bleeding in hereditary hemorrhagic telangiectasia.
Patients with anemia (Hgb <11) at baseline (n = 104) had an increase in mean Hgb after starting bevacizumab, from 8.6 g/dL to 11.9 g/dL at 3 months and up to 12.5 g/dL at 12 months.
The mean epistaxis event went down with treatment from 6.8 at baseline to ≤3.5 during therapy.
For patients who needed RBC transfusions before therapy, transfusion requirements dropped from a median of 8 units in the 6 months before therapy to a median of 0 units in the first 6 months post-therapy.
RBC transfusion and iron infusion requirements continued to improve with ongoing bevacizumab treatment and improvement occurred regardless of baseline bleeding severity.
Requirements for RBC transfusions and iron infusions dropped by 86% and 66%, respectively, with bevacizumab therapy from what they were pretreatment.