 Autosomal dominant disorder most often caused by mutations involving the PTPN11 gene and less frequently NF1 or KRAS genes.
Autosomal dominant disorder most often caused by mutations involving the PTPN11 gene and less frequently NF1 or KRAS genes.
Characterized by nuchal hygromas and hydrops.
Ultrasound reveals hypertelorism, pulmonary stenosis or hypertrophic cardiomyopathy.
Noonan syndrome (NS) is a genetic disorder that may present with mildly unusual facial features, short height, congenital heart disease, bleeding problems, and skeletal malformations.
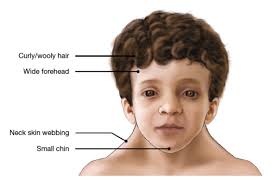
Its facial features include widely spaced eyes, light-colored eyes, low-set ears, a short neck, and a small lower jaw.
Heart problems may include pulmonary stenosis.
The breast bone may either protrude or be sunken, while the spine may be abnormally curved.
Intelligence is often normal.
Complications of NS can include leukemia.
Mildly unusual facial features, short height, congenital heart disease, bleeding problems, skeletal malformations.
Complications: Leukemia
Present at birth
Genetic mutation is autosomal dominant.
Differential diagnosis: Cardiofaciocutaneous syndrome, Turner syndrome, Costello syndrome, neurofibromatosis type 1.
Prognosis depends on the severity of heart problems.
Frequency: 1 in 1000 (1 in 2,000 severe disease)
Noonan syndrome is a type of RASopathy, the underlying mechanism for which involves attenuation of the RAS/MAPK cell signaling pathway.
The diagnosis may be suspected based on symptoms, medical imaging, and blood tests.
Confirmation may be achieved with genetic testing.
No cure for NS is known.
Treatment is based on the symptoms and underlying problems, and extra support in school may be required.
Growth hormone therapy during can increase an affected person’s final height.
Long-term outcomes typically depend on the severity of heart problems.
An estimated 1 in 1,000 people are mildly affected by NS, while about 1 in 2,000 have a more severe form of the condition.
Males appear to be affected more often than females.
The most common signs leading to the diagnosis of Noonan syndrome are unique facial characteristics and musculoskeletal features.
The facial characteristics are most prominent in infancy, becoming less apparent with age in many people with Noonan syndrome.
Characteristic features of Noonan syndrome: a large head with excess skin on the back of the neck, low hairline at the nape of the neck, high hairline at the front of the head, triangular face shape, broad forehead, and a short, webbed neck.
Hypertelorism, widely set eyes, is a defining characteristic, present in 95% of people with Noonan syndrome.
Hypertelorism may be accompanied by epicanthal folds, ptosis, proptosis strabismus, nystagmus and refractive visual errors.
The nose may be small, wide, and upturned.
The development of the ears and auditory system may be affected resulting in low-set ears in over 90% of cases, backward-rotated ears in over 90%, thick helix of outer rim of ear in over 90%, and incomplete folding of ears, chronic otitis media and hearing loss.
The mouth may also be affected in NS with a deeply grooved philtrum in over 90%, micrognathia, high arched palate, articulation difficulties which can lead to dental problems, and poor tongue control may be observed.
Skin signs and symptoms in Noonan syndrome include lymphedema, keloid formation, excessive scar formation, hyperkeratosis, pigmented nevi and connective tissue disease.
Abnormalities in the limbs and extremities may occur in Noonan syndrome: bluntly ended fingers, extra padding on fingers and toes, edema of the back of hands and tops of feet, and cubitus valgus of the elbows.
The final adult height of individuals with Noonan syndrome is about 161–167 cm in males and 150–155 cm in females, which approaches the lower limit of normal.
Spinal abnormalities may be present up to 30% of cases and may require surgery to correct.
Other musculoskeletal manifestations in Noonan syndrome: are associated joint contractures, joint hypermobility, winging of the scapula, scoliosis, breast bone prominence (pectus carinatum), breast bone depression (pectus excavatum), hypotonia which may lead to lordosis due to poor abdominal muscle tone.
Noonan syndrome is the second most common syndromic cause of congenital heart disease.
50-70% of individuals with NS are born with some form of congenital heart defect, with pulmonary valvular stenosis being the most common (50–60%).
Other heart defects in NS that occur include hypertrophic cardiomyopathy (12–35%), ventricular septal defects (5–20%), and atrial septal defects (10–25%).
Restrictive lung function has been reported in some people.
A number of diverse gastrointestinal (GI) symptoms have been associated with Noonan syndrome, including swallowing difficulties, low gut motility, gastroparesis, intestinal malrotation, and frequent or forceful vomiting.
These digestive issues may lead to decreased appetite, failure to thrive from infancy to puberty in 75% of case and occasionally the need for a feeding tube.
In some males testicles do not descend.
Lymphatic anomalies including webbed neck and Lymphedema may present in people with Noonan syndrome.
Bleeding disorders have been associated with Noonan syndrome include platelet dysfunction, blood clotting disorders, partial deficiency of factor VIII, partial deficiency of factor XI, partial deficiency of factor XII, and an imbalance of plasminogen activator inhibitor type-1 (PAI-1) and tissue plasminogen activator (t-PA) activity, association with Von Willebrand disease, amegakaryocytic thrombocytopenia, prolonged activated partial thromboplastin time, and combined coagulation defects.
Patients with NS exhibit a broad range of cognitive abilities, typically ranging from mild intellectual disability to completely normal intelligence.
Most patients have normal IQ levels (70-120), while around 20% may have cognitive impairment.
Occasionally, Chiari malformation (type 1), may occur, which can lead to hydrocephalus.
Seizures may occur.
Mutations in the Ras/mitogen activated protein kinase signaling pathways are known to be responsible for about 70% of NS cases.
Individuals with NS have up to a 50% chance of transmitting it to their offspring.
However, while 30-75% of cases show a noticeable inheritance from one of the parents, the rest are caused by de-novo genetic mutations occurring for the first time in the affected individual.
The fact that an affected parent is not always identified for children with NS.
Several genes are involved in the genetic etiology of NS, with the key ones being PTPN11 accounting for 50% of genetically diagnosed cases, SOS1 responsible for 10-13% of cases, and RAF1 or RIT1 – each contributing to an additional 5% of cases.
Mutations in the PTPN11 gene are associated with an increased tendency for pulmonary stenosis or leukemia, while mutations in the SOS1 gene are linked to relatively normal development and stature compared to other NS cases.
About 15-20% of NS cases remain genetically undiagnosed.
Heterozygous mutations in NRAS, HRAS, BRAF, SHOC2, MAP2K1, MAP2K2, and CBL have also been associated with a smaller percentage of NS and related phenotypes.
Diagnosing of NS is based on the clinical symptoms presented by the individual, with confirmation of the diagnosis through molecular genetic tests to identify the specific genetic change leading to the condition.
Identification of 14 causative genes, however, the absence of a known mutation will not exclude the diagnosis, as more, as-yet-undiscovered genes can cause NS.
The diagnosis of NS is based on clinical features.
An increase in hypertrophic cardiomyopathy is seen in people with a mutation of KRAS and an increased risk of juvenile myelomonocytic leukemia exists for a mutation of PTPN11.
Prenatal features that might lead physicians to consider a diagnosis of Noonan syndrome include cystic hygroma, increased nuchal translucency, pleural effusion, and edema.
Differential diagnosis
Turner syndrome has similarities with renal anomalies and developmental delay, Turner syndrome is only found in females and often expresses differently. In Turner syndrome, there is a lower incidence of developmental delays, left-sided heart defects are constant and the occurrence of renal abnormalities is much lower.
Watson syndrome – Watson Syndrome has a number of similar characteristics with Noonan’s Syndrome such as short stature, pulmonary valve stenosis, variable intellectual development, and skin pigment changes. Cardiofaciocutaneous (CFC) syndrome – CFC syndrome is very similar to Noonan’s Syndrome due to similar cardiac and lymphatic features. However, In CFC syndrome intellectual disability and gastrointestinal problems are often more severe and pronounced.[37][39]
Costello syndrome -has overlapping features with Noonan’s Syndrome. However, the conditions can be distinguished by their genetic cause.
Neurofibromatosis 1 (NF1)
Williams syndrome
Treatment:
Management guidelines, divided by systems, including general, developmental, dental, growth and feeding, cardiovascular, audiological, haematological, renal and skeletal, that account for actions to be taken at diagnosis, after diagnosis and if symptomatic.
Noonan syndrome is a genetic condition affecting multiple organ systems, and requires a comprehensive, multidisciplinary approach to management.
There is no cure for Noonan syndrome as it is a genetically inherited disease.
Management is targeted toward symptomatic improvement and supportive care, to address multiple organ systems.
Patients require systematic evaluation of multiple organ systems:
Cardiovascular Assessment-
Echocardiography and electrocardiography are essential, and a cardiac evaluation is needed every 5 years.
The most common cardiac issues are pulmonary stenosis and hypertrophic cardiomyopathy
Evaluations
Ophthalmologic examination for vision problems and strabismus Audiologic assessment for hearing loss Renal ultrasound to assess kidney structure Coagulation studies to evaluate bleeding disorders Developmental assessment for intellectual and motor delays Chest and spine imaging for skeletal abnormalities
Growth hormone therapy may be considered for short stature
Surgical Interventions In males with cryptorchidism, orchiopexy should be performed when the child is approximately 1 year old if the testes have not descended.
Cardiac surgery may be required for severe congenital heart defects
Correction of skeletal abnormalities like pectus deformities when indicated
Hearing tests and ophthalmic exams are appropriate throughout childhood
Management of lymphedema.
Treatment of bleeding disorders, often related to factor XI deficiency.
The prognosis varies significantly based on the severity of manifestations.
Many patients can have normal lifespans with appropriate management, though some may experience complications including:
Cardiac abnormalities requiring ongoing medical care.
Developmental delays with cognitive deficits.
Bleeding disorders requiring medical management.
Genetic counseling is recommended for families, given the autosomal dominant inheritance pattern with a 50% risk of transmission to offspring.
Speech therapy if speech and articulation issues present
Physical therapy and occupational therapy for gross- and fine-motor delays.
Hypotonia and motor difficulties often impact handwriting.
The lifespan of people with Noonan’s syndrome can be similar to the general population, however, it can be associated with several health conditions that can contribute to mortality.
The greatest contributor to mortality in individuals with Noonan syndrome is complications of cardiovascular disease.
Prognosis is therefore largely dependent on the presence or absence of cardiac disease.
Noonan syndrome with hypertrophic cardiomyopathy is associated with increased mortality.