 The NLRP3 inflammasome
The NLRP3 inflammasome
NOD-, LRR- and pyrin domain-containing protein 3.
NLRP3 is an intracellular sensor that detects a broad range of microbial activities, endogenous danger signals and environmental irritants, resulting in the formation and activation of the NLRP3 inflammasome.
The inflammasome is a multi-protein complex consisting of a sensor, an adapter, and an effector, that when activated, modulates caspases which cleave cytokines and result in an inflammatory signaling response.
Assembly of the NLRP3 inflammasome leads to caspase 1-dependent release of the pro-inflammatory cytokines IL-1β and IL-18, as well as to gasdermin D-mediated pyroptotic cell death.
The NLRP3 inflammasome is a critical component of the innate immune system.
The NLRP3 inflammasome mediates caspase-1 activation and the secretion of proinflammatory cytokines IL-1β/IL-18 in response to microbial infection and cellular damage.
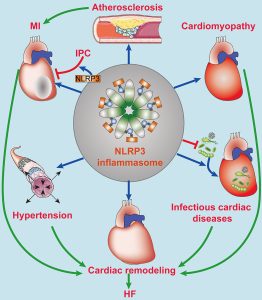
The abnormal activation of the NLRP3 inflammasome has been linked with several inflammatory disorders, which include cryopyrin-associated periodic syndromes, Alzheimer’s disease, diabetes, and atherosclerosis.
The NLRP3 inflammasome is activated by diverse stimuli: molecular and cellular events, including ionic flux, mitochondrial dysfunction, and the production of reactive oxygen species, and lysosomal damage
NLRP3 Inflammasome has a key role in antiviral responses.
NLRP3inflammasome is an oligomeric complex comprised of the NOD-like receptor NLRP3, the adaptor ASC, and caspase-1.
This complex is crucial to the host’s defense against microbes as it promotes IL-1β and IL-18 secretion and induces pyroptosis.
NLRP3 recognizes pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) generated during viral replication that triggers the NLRP3 inflammasome-dependent antiviral immune responses and facilitates viral eradication.
Pattern recognition receptors (PRRs) recognize viral infections and, subsequently, trigger antiviral immune responses.
Several viruses have evolved strategies to evade the immune system by targeting the NLRP3 inflammasome.
Pattern recognition receptors (PRRs) include Toll-like receptors (TLRs), retinoic acid-inducible gene-I (RIG-I) like receptors (RLRs), and DNA sensors such as cyclic GMP-AMP synthase (cGAS), sense different pathogen-associated molecular patterns (PAMPs), and damage-associated molecular patterns (DAMPs) derived from invading viruses.
PRRs can induce the activation of two different transcription factor-mediated pathways, IRF3 and NF-κB.
IRF3 mediates the secretion of type I interferons (IFNs), which lead to the activation of the JAK-STAT pathway and the expression of interferon-stimulated genes (ISGs).
Initiates both the production of proinflammatory factors, such as tumor necrosis factor (TNF)-α and interleukin (IL)-6, as well as the initiation of inflammasome priming.
Some PRRs recruit apoptosis-associated speck-like protein and caspase-1 to form the inflammasome—a multimeric platform of proteins that initiates inflammation as well as some forms of cell death.
Among all the inflammasomes NLRP3 inflammasome is the most extensively studied and it plays an important role in both inflammation and antiviral responses.
NLRP3 inflammasome activation during a viral infection.
There is immune evasion mechanisms of viruses that target the NLRP3 inflammasome.
NLRP3 inflammasome are vital in the host antiviral immune responses.
Several viruses, such as Influenza A and West Nile virus (WNV), tend to induce an appropriate and early phase activation of the NLRP3 inflammasome.
The NLRP3 inflammasome can be activated by sensing viral components as well as cytosolic danger signals, such as mitochondria injury, protein aggregates, and aberrant ion concentrations, all of which can be caused by a viral infection.
NRLP3 inflammasome activation requires two steps: The priming step, is induced by pattern recognition receptors (PRRs) or TNFR activation.
NRLP3 inflammasome activation leads to the activation of NF-κB and promotes the expression of NLRP3, pro-IL-1β, and pro-IL-18.
The second step is triggered by stimuli that emerge during infections, tissue damage, or metabolic imbalances: ATP, pore-forming toxins, crystalline substances, nucleic acids, and invading pathogens.
Once activated, the NLRP3 inflammasome triggers the auto-cleavage of pro-caspase-1.
Caspase-1 mediates the proteolytic processing of pro-IL-1β, pro-IL-18, and the propyroptotic factor gasdermin D (GSDMD).
GSDMD forms pores in the membrane of infected cells, facilitating the secretion of IL-1β/IL-18 and inducing the inflammation-associated cell death known as pyroptosis.
The secretion of IL-1β subsequently recruits neutrophils to the inflammatory site to aid in the elimination of invading viruses.
Moreover, both IL-1β and IL-18 are responsible for the subsequent induction of the adaptive immune response.
The optimal activation of the NLRP3 inflammasome facilitates the establishment of a host antiviral status.
The abnormal NLRP3 inflammasome activation can also lead to severe pathological injury.
HIV-1 infected microglia are shown to cause NLRP3-associated neuroinflammation.
HCV infection promotes chronic intrahepatic inflammation and liver injury mediated by the NLRP3 inflammasome.
In the rest state, cellular NLRP3 level is low enough to avoid aberrant inflammasome assembly and activation.
Viral infection initiates NF-κB signaling through the activation of pattern recognition receptors-dependent pathways.
RIg-like receptors (RLRs) as cytosolic RNA sensors, detect viral RNA.
TLRs participate in the sensing of IAV, HCV, and adenovirus type 5.
Respiratory syncytial virus (RSV), IAV and human parainfluenza virus (HPIV) activate TLRs in macrophages.
HIV can prime NLRP3 inflammasome transcription in monocyte-derived macrophages.
Pattern recognition receptor (PRRs) induced IFN-β and TNF-α that could, in turn, activate NF-κB and provide the cascade amplification necessary for NLRP3 inflammasome activation.
This response enables the host to defend effectively against viral infections.
Viral Infection Triggered The Activation Step Of NLRP3 Inflammasome
The NLRP3 inflammasome can be activated by both viral components, including RNA and proteins (PAMPs), and danger signals (DAMPs).
NLRP3 inflammasome does not directly interact with viral structures, it is still sensitive to invading viruses and cytosolic danger signals, indicating its complicated mechanisms of sensing invading pathogens.
NLRP3 can sense some pathogen-associated molecular patterns (PAMPs).
DAI/ZBP1, and NLRP3 recognize viral proteins and promotes inflammasome assembly.
ThE viral protein sensor DAI/ZBP1 is critical to the induction of NLRP3 inflammasome-mediated apoptotic and necroptotic cell death.
NLRP3 inflammasome is usually associated with sensing cytosolic danger signals referred to as DAMPs.
Intact viruses, also viral components, including internalized or genomic DNA, dsRNA, ssRNA, and even poly(I:C), could directly activate the NLRP3 inflammasome and induce IL-1β secretion in macrophages.
During infection, viruses cause a series of changes in cellular status:
lysosomal maturation, aberrant ion concentrations, mitochondria damage, and the accumulation of misfolded protein aggregates, all of which are recognized as danger signals by the host and lead to the activation of the NLRP3 inflammasome.
Lysosomal changes lead to the leaking of catalytically active cathepsin B, and the subsequent generation of reactive oxygen species (ROS), which, in turn, activates the NLRP3 inflammasome.
Adenovirus type 5 induces the disruption of endosomal membranes and the release of cathepsin B, thereby activating NLRP3.
A balanced ionic concentration is crucial to maintain cellular homeostasis within cells, and when this homeostasis is disrupted, the NLRP3 inflammasome will sense danger signals and become activated.
Potassium efflux is a activator of the NLRP3 inflammasome.
HCV infection induces potassium efflux in macrophages, thus leading to the maturation of pro-IL-1β.
Viroporins are small, highly hydrophobic proteins derived from viruses, which interact with membranes to modify the host cell’s permeability to ions or other small molecules.
Viroporins are observed to localize to the Golgi apparatus and other cytoplasmic structures during viral infection.
Viroporins activate the NLRP3 inflammasome by inducing different ionic fluxes.
Other viral proteins, cause the flux of calcium from intracellular storages to the cytosol, which is indispensable for NLRP3 activation.
The disturbance of ionic concentrations leads to mitochondria damage and the production of reactive oxygen species potentiating NLRP3 inflammasome activation.
Mitochondria damage is also a crucial activator of the NLRP3 inflammasome.
The accumulation of misfolded protein aggregates activates signal of the NLRP3 inflammasome:Alzheimer’s disease is characterized by the accumulation of amyloid-β peptide
SARS-CoV forms intracellular aggregates through the valine residue at position 77, and induces endoplasmic reticulum stress and lysosomal damage, resulting in NLRP3 inflammasome activation.
Viral infections alter the plasma membrane integrity and ionic efflux, leading to programmed cell death and induce the secondary activation of NLRP3 inflammasome.
The process of viral replication causes lytic cell death and subsequent potassium efflux, which provides the second signal for NLRP3 inflammasome activation.
Optimal activation of host immunity is crucial for the elimination of invading viruses.
However, viruses have evolved strategies to evade immune responses by limiting the activation of the NLRP3 inflammasome.
Some viruses have been reported to suppress NLRP3 inflammasome activation to circumvent innate immunity and facilitate viral replication.
Viruses can inhibit both the assembly and the activation of the NLRP3 inflammasome through direct or indirect interactions.
Measles virus and paramyxovirus V protein, and influenza virus NS1 protein inhibit NLRP3 inflammasome activation by interacting with NLRP3, decreasing the secretion of IL-1β accordingly.
NLRP3 ubiquitination and protein degradation is a key regulatory mechanism for NLRP3 inflammasome activation.
Both the NLRP3 inflammasome activation and the subsequent inflammation play significant roles in defending against viral infections.
Aberrant NLRP3 inflammasome activation or chronic inflammation can also lead to severe pathological injury.
The activation of the NLRP3 inflammasome and its associated inflammation is a double-edged sword for host to defense viral infection.