Niemann–Pick disease (NP), also known as acid sphingomyelinase deficiency, is a group of rare genetic diseases.
These are inherited metabolic disorders in which sphingomyelin accumulates in lysosomes in cells of many organs.
NP types A, A/B, and B are cause by mutations in the SMPD1 gene, which causes a deficiency of a acid sphingomyelinase (ASM).
NP type C is now considered a separate disease, as SMPD1 is not involved, and there is no deficiency in ASM.
Type C NP disease is a rare, progressive, debilitating, and prematurely fatal, autosomal recessive lysosomal storage disorder, with an incidence of one case per hundred thousand persons.
These disorders involve the dysfunctional metabolism of sphingolipids, which are fats found in cell membranes.
They can be considered as a kind of sphingolipidosis, which is included in the larger family of lysosomal storage diseases.
Neimann-Pick disease type C is a lysosomal storage disorder of cellular cholesterol trafficking caused by mutations in NPC 1, occurring in 95% of patients with the disorder, or NPC 2 occurring in 5%.
Symptoms are related to the organs in which sphingomyelin accumulates.
It’s spectrum of disease spans rapid, fatal, neurologic regression in infancy to late infantile, juvenile, and adult onset forms, characterized by splenomegaly, supranuclear gaze palsy and neurologic features, such as cerebellar ataxia, dysarthria, and progressive dementia.
NP disease manifests with systemic, psychiatric, and neurologic symptoms.
NP is associated with many neurologic impaired functions.
Enlargement of the liver and spleen may cause reduced appetite, abdominal distension, and pain.
Enlargement of the spleen may also cause thrombocytopenia.
The accumulation of sphingomyelin in the central nervous system results in unsteady gait, slurring of speech, and difficulty swallowing.
Basal ganglia dysfunction causes abnormal posturing of the limbs, trunk, and face.
Within upper brainstem disease results in impaired voluntary rapid eye movements-supranuclear gaze palsy.
More widespread disease causes gradual loss of intellectual abilities, causing dementia and seizures.
Bones also may be affected, with enlarged bone marrow cavities, thinned cortical bone, or a distortion of the hip bone called coxa vara.
Sleep-related disorders also occur with sleep inversion, sleepiness during the day and wakefulness at night.
Gelastic cataplexy, the sudden loss of muscle tone when the affected patient laughs, can occur.
Niemann–Pick disease has an autosomal recessive pattern of inheritance, which means both copies, or both alleles of the gene, must be defective to cause the disease.
Mutations in the SMPD1 gene cause Niemann–Pick disease types A and B produce a deficiency in the activity of the lysosomal enzyme acid sphingomyelinase, that breaks down the lipid sphingomyelin.
Mutations in NPC1 or NPC2 cause Niemann–Pick disease, type C (NPC), which affects a protein used to transport lipids.
Type D originally was separated from type C to delineate a group of patients with otherwise identical disorders who shared a common Nova Scotian ancestry.
Most often, the parents of a child with an autosomal recessive disorder are carriers: they have one copy of the altered gene, but are not affected because the other copy produces the enzyme.
If both parents are carriers, each pregnancy has a 25% chance of producing an affected child.
Genetic counseling and genetic testing are recommended for families who may be carriers of the disease.
Harmful quantities of fatty substances, or lipids, accumulate in the spleen, liver, lungs, bone marrow, and brain.
In the classic infantile type-A variant, a missense mutation causes complete deficiency of sphingomyelinase.
Sphingomyelin is a component of cell membrane including the organelle membranes.
The enzyme deficiency blocks degradation of lipid, resulting in the accumulation of sphingomyelin within lysosomes in the macrophage-monocyte phagocyte lineage.
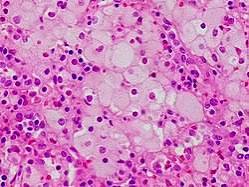
Cells that are affected enlarge, sometimes up to 90 μm in diameter, secondary to the distention of lysosomes with sphingomyelin and cholesterol.
Bone marrow histology shows lipid-laden macrophages in the marrow and “sea-blue histiocytes”.
Small vacuoles give the cytoplasm a foamy appearance.
Diagnosis:
For type A and B, levels of sphingomylinase can be measured in the blood sample.
To diagnose type C, a skin sample can help determine whether the transporter is affected via the Filipin test.
The Filipinos test detects build-up of unesterified cholesterol via fluorescent staining.
The four types of Niemann–Pick disease are divided into categories.
Patients with acid sphingomylinase deficiency are classified into types A and B.
Type A patients exhibit hepatosplenomegaly in infancy and profound central nervous system involvement, and are unable to survive beyond two years of age.
Type B patients also show hepatosplenomegaly and pathologic alterations of their lungs, but usually without the involvement of their central nervous system.
Some patients can develop significant life-threatening complications, including liver failure, hemorrhage, oxygen dependency, pulmonary infections, splenic rupture, coronary arterial or valvular heart disease.
In a longitudinal natural history study, nearly 20% of the patients die.
For those classified into type C, they may have mild hepatosplenomegaly, but their central nervous system is profoundly affected.
Type C is the most common form of the disease.
Type C2 is a rare form of the disease.
Two poorly characterized forms of Niemann–Pick disease have also been described as types E and F.
No specific treatment is known for type A.
With type B, keeping cholesterol levels down to normal levels.
Bone marrow transplant has been tried for type B.
Treatment
No cure exists for Niemann-Pick disease, but supportive care can help manage symptoms.
For Niemann-Pick disease type B, olipudase alfa-rcpc (Xenpozyme) is an enzyme replacement for missing or low levels of the sphingomyelinase enzyme.
Olipudase alfa-rcpc enzyme replacement may help with lung problems and breathing and lessen the size of the liver and spleen.
Olipudase alfa-rcpc enzyme may help height growth in children.
It does not help with nerve-related symptoms.
Olipudase alfa-rcpc is given intravenously every two weeks.
For people with Niemann-Pick disease type C who have mild to moderate nerve symptoms, Miglustat is approved for neurological symptoms.
Miglustat is a pill that’s taken 1 to 3 times a day, which slow the worsening of nerve symptoms such as problems with hearing, swallowing, walking, and may help slow changes in mental health and learning and memory.
The use of the amino acid analog N-acetyl leucine (NALL) showed significant improvement in ataxia scores.
Infantile neurovisceral Niemann Pick disease (Type A ASMD) is usually fatal before 3 years of age.
In Type B, severity is highly variable, and many patients live well into adulthood and may reach a normal lifespan.
Diagnoses of Type B have been made in the 7th decade of life.
Type C also has a highly variable prognosis.
The incidence among Ashkenazi Jews is estimated to be about one in 40,000 for type A of Niemann–Pick disease.
The incidence of both Niemann–Pick disease types A and B in all other populations is estimated to be one in 250,000.
The incidence of Niemann–Pick disease type C is estimated to be one in 150,000.
At present there is no cure.
The loss of myelin in the central nervous system is considered to be a main pathogenic factor.
The expression of myelin gene regulatory factor (MRF) has been shown to be significantly decreased.
MRF is a transcription factor of critical importance in the development and maintenance of myelin sheaths, and a perturbation of oligodendrocyte maturation and the myelination process may an underlying mechanism of the neurological deficits.