 A low grade cutaneous lymphoma with skin-homing CD 4 positive T cells.
A low grade cutaneous lymphoma with skin-homing CD 4 positive T cells.
Unknown etiology.
Incidence estimated at 5.42 per million per year and the incidence has increased annually by 1.34% over the past two decades.
The median age at diagnosis is 55 to 60 years.
There is a male to female ratio of two to one.
Constitutes 4% of non-Hodgkin’s lymphoma, but represents the most common extra nodal lymphoma.
The most common primary cutaneous lymphoma, comprising nearly 50% of all cases.
Accounts for 50% of all primary cutaneous lymphomas and 50% of all cutaneous T cell lymphomas.
Increasing incidence of 6.4 per million.
The most common form of T-cell cutaneous lymphoma.
Classified as mycosis fungoides: classic mycosis fungoides (88.6%) and three variants.
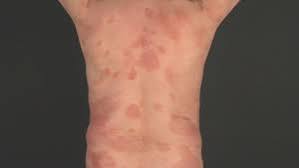
Classic MF is slowly progressive disease that occurs primarily are non-exposed areas of skin and appears as cutaneous patches, then cutaneous plaques, and later as exophytic or ulcer related tumors.
Pruritis is present in 80% of patients with MF.
Extracutaneous MF occurs, rarely can involve lymph nodes, spleen, liver, lungs, and G.I. tract.
Indolent primary cutaneous T-cell Lymphoma.
Involves clonal liberation of malignant transformation of skin homing CD 4 memory T cells.
Differential diagnosis includes: eczema, psoriasis, drug reactions, contact dermatitis and other cutaneous cell and T cell lymphomas.
Can affect people of any gender, race and age.
More common in men.
Disproportionately affects black men between the ages of 55 and 60.
Patients with mycosis fungoides are at increased risk for developing other cancers and should be screened for second primary and hematologic malignancies.
MF is usually associated with an indolent clinical course and intermittent, stable, or slow progression of lesions.
Extra cutaneous involvement of lymph nodes, blood or less commonly other organs or large cell transformation may be seen in advanced stage disease.
A conversion to Sezary syndrome characterized by generalized erythroderma and circulating cerebriform cells in the peripheral blood.
Tendency to develop skin patches and plaques.
Presentation with eczematous dermatitis in sun protected sites such as the trunk, axilla and buttocks.
Lesions may be ill or well defined, and manifest as erythematous pruritic patches, plaques or a large necrotic tumors.
Typically lesions initially appeared in non sun exposed areas and evolve from patches to plaques and then tumors.
Skin tumors can become very large and may be mushroom shaped accounting for the term fungoides.
Premycotic erythematous scaling lesions can wax and wane for years.
May clinically resemble dermatitis, eczema, or psoriasis.
May be associated with alopecia and generalized erythroderma.
In the past phase of disease involvement begins in the trunk, pelvis, and proximal extremities.
Patch-plaque disease stages IA, Ib, and IIa have a survival of more than 12 years.
Patients with erythroderma , stages mIIb/III have a median survival of approximately 4 years.
Patients with stage IV, which includes those patients with lymph node or visceral involvement, have a median survival of less than 3 years (Demerred MF, Vonderheld EC).
Most agents available for advanced disease have a response rate of 30% overall, especially after bexarotene.
It may take up to 3 agents before an active drug is identified.
Diagnosis may take up to years as the disease is often confused with psoriasis or eczema.
Diagnosis requires the presence of erythematous scaling lesion in sun-protected areas and can wax and wane for years.
Diagnosis is made with skin biopsy of patches or plaques, revealing a band like lymph infiltrate in the superficial dermis with atypical lymphocytes, extending into the epidermis.
Tumor lesions and neoplastic lymphoid infiltrate extends into the dermis.
In 80% of cases CD 4/CD eight type immunophenotype predominates in the lymphoid infiltrate.
The diagnosis is supported by absence of antigen of normal T cell such as CD2, CD3, CD5 and CD7.
Biopsies of the lesions will reveal atypical lymphocytes infiltrates of the epidermis, or dermis if the disease is advanced.
Pautrier’s microabscess are pathognomonic but not common.
85% of patients present with only patch or plaque disease T1-2N0-1M0B0, i.e. stage IA and IB.
May spread to regional lymph nodes, blood, central lymph nodes,and visceral organs.
Prognosis poor for advanced disease.
As disease progresses the T-cell lymphocyte population is destroyed and the patients become immunocompromised.
Mortality often due to infections from necrosis and ulcerations of lesions.
Typically have CD4 helper cell phenotype.
Patients have a predilection for increased infections secondary to cellular immunodeficiency with gradual CD8+ cytotoxic population being depleted associated the malignant clone grows.
Patients are predisposed to infections because of the disruption of the normal skin barrier.
Infections, frequently nosocomial type, are the leading cause of death in 27-60% of patients.
If the disease is confined to less than 10% of the body surface area, no decrease in life span is expected.
Median survival for patients with skin involvement of 10% or more is 10.7 years.
Life expectancy is not altered in clinical stage IA disease.
5-year overall survival ranges from nearly 100% for stage IA to less than 10% for stage IVB.
Patients with plaque/patch disease, T1 disease, have a survival similar to the general population.
Patients with a solitary patch or a few patches are typically treated with topical therapy.
Patients with extensive patches and plaques (T2), tumors (T3), and erythroderma (T4), have a median survival approximately 12, 3, and 5 years, respectively (Kim YH).
With progression of the disease there is the development of atypical lymphocytes which lose their affinity for the epidermis and infiltrate the dermis, blood, and lymph nodes.
Median survival for stage IV disease is only 1.5 years.
Large cell transformation within 2 years of diagnosis is associated with poor prognosis.
Large cell transformation frequently refractory to biological response modifiers retinoids and interferon alfa.
Early stage disease often responds to skin directed treatments which includes topical steroids, retinoids, nitrogen mustard and photo therapy.
Topical therapy with chemotherapy, phototherapy and radiation therapy may promote later development of skin cancer.
Interferon and retinoids have response rates of around 50%.
Patients with erythroderma and those with positive lymph nodes have median survival rates of 3.3 and 3.7 years, respectively.
97% of patients with late stage mycosis fungoides or Sezary syndrome are seropositive for cytomegalovirus compared to 57% of healthy bone marrow donors.
In later stages can manifest with peripheral lymphadenopathy and progress the widespread extracutaneous involvement.
Lung involvement is the most common site of visceral involvement and is a common finding at autopsy.
Mycosis fungoides treated in a variety of ways including: sunlight, ultraviolet light, topical steroids, topical and systemic chemotherapies, local superficial radiotherapy, the histone deacetylase inhibitor vorinostat, total skin electron beam radiation, photopheresis and systemic therapies such as interferons, retinoids, rexinoids or biological therapies.
The above treatments are often used in combination.
Treatments varies with access to therapies, the stage of the disease, exposure to prior therapies.
An incurable disease, but quality of life is a major objective.
Patients experience prolonged periods of disease-control.
Nloxone lotion, a topical opioid receptor competitive antagonist can be used as a treatment for pruritus in cutaneous T-cell lymphoma.
Treatments include photophoresis, denileukin diftitox, bexarotene, and histone deacetylase inhibitor.
Brentuximab has an overall response rate of 70% in refractory/advanced mycosis fungoides and Sezary syndrome.
Brentuximab indicated for the treatment of adult patients with primary cutaneous anaplastic large cell lymphoma (pcALCL) or CD30-expressing mycosis fungoides (MF) who have received prior systemic therapy.
FDA approval was based in part on the clinical trial results from ALCANZA, a global, phase 3, randomized study evaluating methotrexate or bexarotene.