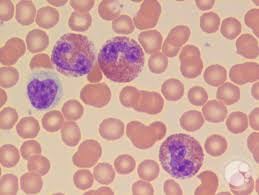
 Lymphocytic variant hypereosinophilic syndrome (L-HES) is a rare, indolent subtype of hypereosinophilic syndrome.
Lymphocytic variant hypereosinophilic syndrome (L-HES) is a rare, indolent subtype of hypereosinophilic syndrome.
It consists of clonaland reactive hematological processes.
Persistent high eosinophil levels are driven by an underlying clonal population of abnormal T cells.
These “aberrant” T cells produce excessive amounts of [interleukin-5 (IL-5) the primary cytokine that triggers eosinophil production and survival.
Aberrant T‑cell clones (most often CD4⁺ Th2‑polarized cells) secrete eosinophilopoietic cytokines that stimulate bone marrow CFU‑Eos, leading to marked but reactive (non‑clonal) eosinophilia.
Eosinophilia is driven by production of cytokines, primarily Interleukin-5, but sometimes Interleukin 4, and possibly Interleukin 13, by an aberrant dysregulated clonal T cell population.
This T cell population commonly lacks CD3 marker and express CD4 on flow cytometry.
IL-5 promotes eosinophil differentiation, chemotaxis activation, and survival and functions as a stimulant upstream of both eosinophil and IgE production: approximately 70% of patients with LHE have an elevated IGE level.
Unlike the myeloid which is a bone marrow disorder, L-HES is an immune-driven disorder where clonal T cells act as the driver for eosinophilia.
The most common abnormal cell profile is CD3−CD4+, meaning the T cells have lost their standard surface CD3 marker but remain positive for CD4.
TCR gene rearrangement studies often confirm clonality; cytogenetic abnormalities may involve chromosomes 6q, 10p, 16q, or trisomy 7 in the aberrant T‑cell population.
Clinical Presentation of L-HES
Patients meet HES criteria: eosinophils typically ≥1.5 × 10⁹/L with eosinophil‑mediated organ involvement, after exclusion of secondary causes and myeloid neoplasms.
Laboratory: often very elevated IgE, sometimes polyclonal hypergammaglobulinemia; eosinophil counts vary widely and can be extremely high or modestly elevated.
Disease is usually indolent but chronic; common manifestations are:
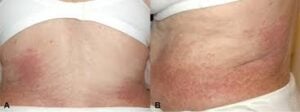
Cutaneous: pruritus, eczematous or urticarial rash, angioedema.
Predominantly affects the skin (>80% of cases), causing itchy rashes-eczema, urticaria, pruritus and episodic angioedema.
Respiratory: asthma‑like symptoms, cough, wheeze.
GI and soft‑tissue involvement.
Rheumatologic symptoms; less often cardiac or neurologic disease than in some other HES variants.
Can involve the lungs by pneumonitis, lymph nodes, and gastrointestinal tract.
Overlap hypereosinophilic syndrome includes eosinophilic disorders that are restricted to a single organ system – eosinophilic esophagitis, eosinophilic fasciitis, and eosinophilic pneumonia, and eosinophilic granulomatosis with polyangiitis.
Secondary Signs: Often associated with high serum immunoglobulin E (IgE) and increased levels of TARC/CCL17.
Diagnosis requires specialized flow cytometry to identify the aberrant T-cell population and T-cell receptor (TCR) gene rearrangement.
Flow cytometry of peripheral blood and often bone marrow to identify aberrant T‑cell populations with the classic Th2 immunophenotype; even small populations (~0.5–1% of lymphocytes) can be clinically meaningful.
TCR gene rearrangement testing to demonstrate clonality and support L‑HES vs reactive eosinophilia; assessment of serum cytokines (IL‑5, sometimes IL‑4/IL‑13) and chemokines (e.g., CCL17) can be supportive but are not required.
Confirm hypereosinophilia and organ involvement; rule out secondary eosinophilia (parasitic infection, drugs, atopy, autoimmune disease, IgG4‑related disease, vasculitides) and myeloid neoplasms (including FIP1L1‑PDGFRA, PDGFRB, FGFR1, JAK2 rearrangements).
Long-term monitoring is essential because these clonal T cells carry an increased risk—roughly 10% to 25%—of transforming into malignant T-cell lymphoma over several years.
Treatment Options
While no single standardized cure exists, treatment focuses on controlling eosinophil-induced organ damage:
Corticosteroids: Usually the first-line therapy.
Systemic glucocorticoids are often first‑line for symptomatic hypereosinophilia but L‑HES may be less steroid‑responsive than idiopathic HES.
Interferon-alpha: Often effective as a second-line steroid-sparing agent and may help reduce the size of the underlying T-cell clone.
Interferon‑alpha has well‑described efficacy in L‑HES due to its anti‑eosinophilic and immunomodulatory effects and can be steroid‑sparing.
Monoclonal Antibodies: Mepolizumab (anti-IL-5) can effectively lower eosinophil counts but generally does not eliminate the underlying abnormal T cells.
Anti‑IL‑5/IL‑5R biologics (e.g., mepolizumab, reslizumab, benralizumab) and JAK‑inhibitors have emerged as effective options in steroid‑refractory or intolerant cases, targeting either IL‑5 signaling or downstream pathways.
Mycophenolate mofetil, cyclosporine, and [JAK inhibitors are emerging alternatives for refractory cases.
Standard HES measures for organ protection (e.g., cardiac monitoring, anticoagulation where indicated) remain essential.
Prognosis and malignant transformation
Most patients have a chronic, indolent course with recurrent or persistent eosinophilia and symptoms but can be controlled with immunomodulatory therapy.
A significant minority (about 10–25%) progress over years to overt T‑cell lymphoma (e.g., angioimmunoblastic T‑cell lymphoma, peripheral T‑cell lymphoma, cutaneous T‑cell lymphoma, or ALK‑negative anaplastic large‑cell lymphoma), at which point disease is aggressive and prognosis poor.
Follow‑up with periodic clinical exam, blood counts, and imaging/lymph node assessment is recommended to detect clonal evolution early.