 Joubert syndrome is a rare autosomal recessive genetic disorder that affects the development of the cerebellum, a part of the brain that controls coordination and balance, and brainstem.
Joubert syndrome is a rare autosomal recessive genetic disorder that affects the development of the cerebellum, a part of the brain that controls coordination and balance, and brainstem.
Affects 1 in 80,000-100,000 newborns.
An X-linked recessive pattern of inheritance.
Males are therefore more commonly affected.
Commonly found in Ashkenazi Jewish, French-Canadians, and Hutterite ethnic populations.
The syndrome is characterized by a group of physical and neurological abnormalities, including:
Abnormal breathing patterns
Poor muscle tone and weakness
Coordination and balance problems
Delayed motor development
Nystagmus
Retinal dystrophy
Intellectual disability
Kidney abnormalities
Liver disease
Skeletal abnormalities
Hormonal abnormalities
Abnormalities in the structure of the brainstem.
Joubert syndrome is one of the many genetic syndromes associated with syndromic retinitis pigmentosa.
This disorder can be caused by mutations in more than 30 genes.
Genetic mutations of primary cilia can disrupt significant signaling pathways during the development of the fetus.
It is a ciliopathy, with a dysfunctional molecular mechanism in the primary cilia structures of the cell, organelles which are present in many cellular types throughout the human body.
The cilia defects adversely affect developmental signaling pathways essential to cellular development.
Mutations in these various genes are known for causing around 60-90% of Joubert Syndrome cases.
It is usually diagnosed in infancy or early childhood.
Symptoms of the Joubert syndrome appear very early in infancy with most children showing delays in gross motor milestones.
The severity of symptoms can vary widely among individuals with the disorder.
Symptom generally fall under the hallmark of cerebellum involvement.
Most common features include ataxia, lack of muscle control, hyperpnea, sleep apnea, abnormal eye and tongue movements, and hypotonia in early childhood.
Malformations such as polydactyly, cleft lip or palate, tongue abnormalities, and seizures may also occur. Developmental delays, including cognitive, are always present to some degree.
Delays in gross motor skills, fine motor skills and speech development are seen in almost all individuals.
Delays can be secondary to low muscle tone as well as impaired motor coordination.
In severe disease hypoplasia of the corpus callosum may be present.
Facial abnormalities such as a broad forehead, arched eyebrows, ptosis, hypertelorism, low-set ears and a triangle shaped mouth may be present.
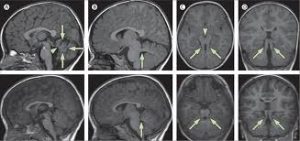
MRI Brain, axial T2 sequence shows enlarged superior cerebellar peduncles and vermian hypoplasia resulting in characteristic “molar tooth” appearance.
There is absence or underdevelopment of the cerebellar vermis and a malformed brain stem (molar tooth sign), both visualized on a transverse view of head MRI scan.
Diagnosis is based on the physical symptoms and genetic testing for mutations.
No cure exists.
Treatment is supportive and aimed at managing the individual symptoms:
It may involve medications, surgery, and physical therapy to improve mobility and coordination.
Developmental delays are usually treated with physical therapy, occupational therapy, and speech therapy interventions, and most children are able to achieve standard milestones, although often at a much later age.
Because the syndrome is associated with progressive worsening for kidneys, the liver and the eyes requiring regular monitoring.