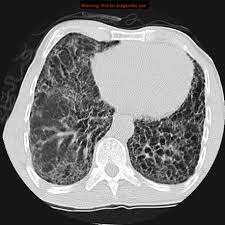
Interstitial lung disease(ILD) is characterized by inflammation and/or fibrosis within the alveolar infestation of the lung.
Approximately 30 to 40% of patients with ILD develop progressive pulmonary fibrosis which causes respiratory failure is associated with the median survival approximately 2 1/2 to 3 1/2 years.
ILD affects approximately 650,000 people in the US.
Estimated prevalence of 179.7 per 100,00 in males and 218.9 per hundred thousand in females.
The prevalence increases with age and is highest in individuals aged 80 to 84 years.
The mean age at diagnosis is approximately 67 to 72 years.
Idiopathic pulmonary fibrosis (IPD) occurs in 198,000 people in the US resulting in 26,000 deaths per year.
Idiopathic pulmonary fibrosis is more common in males and females with an incidence of approximately 3-9 per hundred thousand.
Diffuse parenchymal lung disease involving a large number of conditions, clinical manifestations, imaging and pathological features.
This process has variable outcomes. Each of the individual fibrosing ILDis rare, but collective they effect many patients.
Overall prevalence in the US is 74.3 cases per hundred thousand.
Sarcoidosis and connective tissue disease, and idiopathic pulmonary fibrosis are the most common fibrotic ILD‘s with an estimated prevalence of 30.2, 12.1, and 8.2 cases per hundred thousand, respectively.
Most fibrotic lung diseases have pulmonary alveolar walls infiltrated by various combinations of inflammatory cells, fibrosis, and proliferation of normal alveolar walls cells.
Chronic inflammation increases the thickness of the alveolar pulmonary capillary membrane, which limits the diffusion of oxygen from the alveoli to the blood.
These lung abnormalities predominate in the interstitium and, these disorders are termed interstitial lung disease.
In adults three main patterns of interstitial lung disease exist: usual interstitial pneumonitis, nonspecific interstitial pneumonitis, and lymphoid interstitial pneumonitis (American Thoracic Society).
Hypersensitivity pneumonitis is a form of ILD caused by inhalation of specific antigens, most commonly avian proteins and mold or fungal spores.
The prevalence of hypersensitivity pneumonitis ranges from 1.67 to 2.71 per 100,000, with prevalence increasing with age.
Comprise a group of diffuse parenchymal infiltrative lung disorders classified avoided according to etiological, clinical, radiologic, and histopathologic features.
CT of the chest is the mainstay of a ILD diagnosis.
Phenotyping of the type of ILD depends on assessment of environmental, occupational, or other exposures, such as radiation to the thorax, certain medications, and the evaluation, for the presence of other systemic diseases, including rheumatoid arthritis, and systemic sclerosis.
It is a common phenotype in a broad variety of diseases including: autoimmune, granulomatous, and idiopathic interstitial pneumonias.
Some of the most common forms of ILD such as idiopathic pulmonary fibrosis, are of unknown origin.
Interstitial lung diseases include connective tissue diseases characterized by immune mediated tissue injuries that can involve the lungs.
There is a lack of a diagnostic marker that distinguishes fibrosing interstitial lung disease from idiopathic pulmonary fibrosis.
Connective this tissue disease related ILD usually presents in patients already diagnosed with connective tissue disease.
Up to 90% of patients with systemic sclerosis have radiologic features of interstitial lung disease on high-resolution CT and has the highest prevalence, followed by those of dermatomyositis/polymyositis 20-78%, and rheumatoid arthritis 6.5-33%.
ILD may be the presenting feature, however, accompanied by findings suggestive of an underlying or immune crisis but not sufficient for a diagnosis of connective tissue disease
Patients with connective tissue disease and ILD have substantial mobility and mortality.
The five-year mortality rate is threefold higher in patients with rheumatoid arthritis and scleroderma who have ILD.
When compared with idiopathic interstitial pneumonia is patients with connective tissue disease-ILD more likely to respond to immunosuppressive and have a better prognosis.
ILDs are characterized by restrictive ventilatory defects that is reduce the total lUNG capacity, normal force expiratory volume in 1 second/forced vital capacity FVC ratio would reduce diffusing capacity of the lung for carbon monoxide: however spirometry can be normal and mild disease with mixed obstructive restrictive disorders.
Interstitial pneumonitis is characterized by alteration of the lung architecture by fibrosis.
Refers to a complex group of more than 150 different disorders, which are characterized by inflammation and fibrosis of the pulmonary interstitial and parenchyma.
Interstitial lung abnormalities noted among approximately 70% of adult patients and It is associated with a higher rate of all cause mortality
Usual interstitial pneumonitis is the most common adult form of interstitial lung disease.
Interstitial lung abnormalities associated with older age, smoking, and a restrictive lung deficit.
Patients with ILD may have similar clinical presentations.
Diagnosis requires attention to details of the history, clinical presentation, x-ray findings, and histpathology.
Idiopathic interstitial pneumonias are a subset of disease where a specific ideology is not determined.
May be secondary to drugs, connective tissue diseases, and granulomatous disease that can cause both lung injury and scarring.
Onset of interstitial lung disease is usually during the fifth decade.
Interstitial lung abnormalities are defined as a pattern of increased lung density on chest CT scan in patients with no prior history of interstitial lung disease.
ILD is detected and up to 60% of patients with rheumatoid arthritis on high-resolution CT scan and is clinically significant in 10%, and is the leading cause of illness and death in patients with rheumatoid arthritis.
ILD associated rheumatoid disease shears several characteristics with idiopathic pulmonary fibrosis, including common environmental risk factors, high prevalence of pattern as usual interstitial pneumonia, progressive lung fibrosis, and poor survival.
MUC5B promoter of variance is associated with rheumatoid arthritis-ILD.
Interstitial lung disease associated with decreased lung capacity, decreased exercise capacity, decreased gas exchange, genetic abnormalities, common to patients with familial pneumonia and idiopathic pulmonary fibrosis.
It is suggested that interstitial lung abnormalities may represent early or mild form of pulmonary fibrosis.
The major idiopathic interstitial pneumonias are divided into chronic, smoking related, acute, and subacute diseases.
Idiopathic pulmonary fibrosis is the most common idiopathic interstitial pneumonia, with an estimated incidence of 16.3 per 100,000 individuals.
Connective tissue related interstial lung diseases, sarcoidosis and lymphangiolieomatosis are more common in the middle aged patients.
The risk of lung cancer is increased in patients with ILD.
A metaanalysis of 35 studies estimated the prevalence of lung cancer in patients with idiopathic pulmonary fibrosis at 13.5% with higher rates in men and in smokers.
Patients with ILD and systemic sclerosis have a higher risk of lung cancer.
Idiopathic pulmonary fibrosis is a disease of the elderly.
Many cases occurring in the pediatric population
Nonspecific interstitial pneumonitis has less pulmonary scarring and a better prognosis than usual interstitial pneumonitis, and is generally not seen in infants.
Pediatric ILD is classified on the basis of the location of the primary disease-the airways, that have the alveolus or the interstitium.
In childhood interstitial lung disease impairment in surfactant pathways are important in pathophysiology.
In pediatric patients the diagnosis of interstitial lung disease is considered when has tachypnea, fine pulmonary crackles and hypoxemia.
ILD incidence in children is 0.36 cases per hundred thousand with a male predominance 60:40.
20% of pediatric patients with interstitial lung disease have wheezing, and most cases start with a viral illness.
Three fourths of cases of ILD in children, manifest before the age of one, with the median onset of eight months.
Almost 20% of pediatric cases are associated with a family history of all ILD.
Approximately 6% of children with ILD have lymphoid interstitial pneumonitis seen with autoimmune disease and immuno deficiencies.
Nintedanib is associated with a significant slowing of the decline in the forced vital capacity in patients with interstitial fibrosing lung disease.
Nintedanib is an tyrosine kinase inhibitor.