 Immunodysregulation polyendocrinopathy enteropathy, X-linked (or IPEX) syndrome is a rare disease linked to the dysfunction of the gene encoding transcription factor forkhead box P3 (FOXP3).
Immunodysregulation polyendocrinopathy enteropathy, X-linked (or IPEX) syndrome is a rare disease linked to the dysfunction of the gene encoding transcription factor forkhead box P3 (FOXP3).
Mutations in FOXP3 gene cause the IPEX syndrome.
IPEX syndrome is caused by loss of function variants in FOXP3, which is a master transcriptional regulator that is fundamental to the function of CD4 positive in CD 25 positive regulatory T cells.
Regulatory T cells suppress the activation and effector function of self-reactive and potentially pathogenic lymphocytes.
Loss of function variants in FOXP3 cause quantitative or functional deficiencies in regulate regulatory T cells which lead to immune dysregulation.
The dysregulation can manifest as autoimmune disease and allergic inflammation.
FOXP3 is considered to be the master regulator of the regulatory T cell lineage.
It leads to the dysfunction of CD4+ regulatory T-cells and the subsequent autoimmunity.
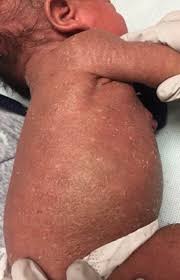
An autoimmune polyendocrine syndrome that manifests with autoimmune enteropathy, psoriasiform or eczematous dermatitis, nail dystrophy, autoimmune endocrinopathies, and autoimmune skin conditions such as alopecia universalis and bullous pemphigoid.
It typically develops in infancy manifesting as dysfunctional regulatory T cells.
Lethal disorder of childhood.
Its classic triad of manifestations: auto immune endocrinopathies typically neonatal type one diabetes and thyroiditis, auto immune enteropathy associated with failure to thrive in severe chronic diarrhea, and eczematous dermatitis.
Symptoms of enteropathy begin in first day of life: diarrhea, vomiting, gastritis, ileus and colitis.
The second hallmark is type 1 diabetes.
Dermatitis the third sign and it can be presented in three forms: eczematiform, ichthyosiform and psoriasiform or combinations of them.
Additional skin manifestations: cheilitis, onychodystrophy and alopecia.
Other findings include : thyroid and renal dysfunction, reduced WBCs and platelets, arthritis, splenomegaly, lymphadenopathy, infections and hematologic disorder is as immune mediated cytopenias, pulmonary disease, hepatitis, renal disease, and infections.
Allergic inflammation can include eczema, food allergies, eosinophilia and elevated total IgE I level.
The disease is marked by a triad of symptoms: intractable diarrhea, T1D, and eczema.
Clinical manifestations include: enteropathy, skin manifestations, endocrinopathy, hematologic abnormalities, infections, autoimmune hemolytic anemia and food allergy.
It is inherited in males via an x-linked recessive manner, as the FOXP3 gene’s, cytogenetic location is Xp11.23.
The FOXP3 gene has 12 exons and it encodes 431 amino-acids.
FOXP3 is a member of the FKH family of transcription factors and contains a proline‐rich (PRR) amino‐terminal domain, central zinc finger (ZF) and leucine zipper (LZ) domains important for protein–protein interactions, and a carboxyl‐terminal FKH domain required for nuclear localization and DNA‐binding activity.
A consequence of malfunctioning FOXP3 expression leads to a defect in Treg production.
Those patients do not have circulating CD4+/CD25+/FOXP3+ Treg cells.
Mutation of FOXP3 leading to expression of malfunctioning protein is often localized in the DNA-binding domain called the forkhead domain.
The truncated protein cannot bind to its DNA binding site and T regulatory lymphocytes development and functioning is impaired.
The absence or dysfunction of regulatory T cells is the cause of autoimmune symptoms.
There are over 70 mutations in FOXP3 gene leading to IPEX syndrome.
FOXP3 can function as both a repressor and a trans‐activator of Treg cells depending on its interactions with other proteins.
FOXP3 expression is characterized by controlling transcription, influencing epigenetic and post-transcriptional modifications.
FOXP3 can change transcription or epigenetic regulation of Treg cells.
FOXP3 is important in the regulation of transcriptional activity/repression in Treg cells.
This autoimmunity called IPEX is an attack from the body’s own immune system against the body’s own tissues and organs.
It has an early age onset of this disease in males and causes severe enlargement of the secondary lymphoid organs, and insulin dependent diabetes.
There is loss of CD4+ CD25+ T regulatory cells, and expression of the transcription factor Foxp3.
Foxp3 decrease is a consequence of unchecked T cell activation, which is secondary to loss of regulatory T cells.
Causes of death in IPEX syndrome include hemorrhage, sepsis, Intractable diarrhea and diabetic complications.
The diagnosis of immunodysregulation polyendocrinopathy enteropathy X-linked syndrome criteria:
Clinical triad: Its classic triad of manifestations: auto immune endocrinopathies typically neonatal type one diabetes and thyroiditis, auto immune enteropathy associated with failure to thrive in severe chronic diarrhea, and eczematous dermatitis.
Family history
Laboratory findings: elevated serum concentration of IgE, eosinophilia, autoimmune anemia and decreased number of FOXP3 Treg cells.
Genetic testing: single-gene testing and multigene panel.
Treatment
Corticosteroids are the first treatment.
TPN
Cyclosporin A and Tacrolimus
Sirolimus
Granulocyte colony stimulating factor
Bone marrow transplant:Allogeneic hematopoietic stem cell transplantation can restore T-cell regulatory function but is associated with a poor outlook.
Rituximab