 A chronic inflammatory disease manifested by ill defined fibroproliferative infiltration of lymphoplasmacytic cells into various organs by IgG4 positive cells.
A chronic inflammatory disease manifested by ill defined fibroproliferative infiltration of lymphoplasmacytic cells into various organs by IgG4 positive cells.
A rare, chronic, relapsing, immune mediated fibroinflammatory disorder with a reported prevalence of 5.3 people per hundred thousand in the US.
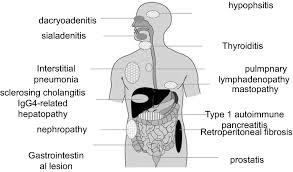
It can affect multiple organs, including the pancreas, salivary glands, lymph nodes, and kidneys, as well as the aorta, retroperitoneum, peripheral nervous system, and CNS.
It is characterized as a multi system fibroinflammatory disorder with infiltration of plasma cells and lymphocytes.
A multi organ immune mediated condition.
A complex disease that is difficult to diagnose.
It is a relapsing, immune related inflammatory condition.
Pathology: Dense polyclonal lymphoplasmacytic infiltrates rich in IgG4 positive plasma cells with fibrosis obliterative phlebitis and tissue eosinophilia.
It is characterized by the development of mass lesions, rich in CD19 positive B cells that may drive inflammation and fibrosis by means of cytokines or indirectly by the activation of pathogenic T cells.
It is characterized by insidious accumulation of immune cells in organs, which generally manifest is diffuse organ enlargement.
Typically IgG4 is anti inflammatory.
Multiorgan involvement is typical and includes the lacrimal glands, salivary glands, pancreas, bile ducts, retroperitoneal tissues, kidneys, and aorta.
Disease flares may lead to progressive organ dysfunction and failure.
Pathogenesis involves antigen driven interactions between B-cell subsets and various CD4 positive and CD8 positive T lymphocytes that drive tissue injury and fibrosis and are associated with all the clonal expansions of plasmablasts and increased levels of serum IGG4.
Histopathologic characteristics include: lymphoblastic infiltrates, rich in IgG4 plasma cells, fibrosis, obliterative phlebitis, tissue eosinophilia, expanded populations of plasmablasts, and increased CD19 positive B cells.
Kidney manifestations include 3 subtypes: 1 obstructive uropathy mainly secondary to retroperitoneal fibrosis; 2 IgG4 related tubulointerstitial nephritis; 3 glomerular lesions particularly membranous nephropathy.
Nephritis has characteristic radiologic features such as bilateral nodular or wedge-shaped lesions showing low contrast-enhancement on CT and T2 weighted MRI.
On biopsy histopathological features include severe tubular atrophy, storiform fibrosis diffuse inflammatory infiltrate rich in plasma cells and eosinophils.
Glucocorticoids may be insufficient with membranous nephropathy; rituximab is associated with lower risk of relapse
Interestingly IgG4 related kidney disease is almost never identified as a single organ disease
C3 and C4 are frequently reduced.
When there is kidney involvement there is frequently other organ manifestations including autoimmune pancreatitis dacryoadenitis and sialodenitis
Termed IgG4-related disease (IgG4RD).
High IgG4 level seen in other inflammatory allergic and neoplastic diseases.
Prevalence is 4.6 cases 100,000.
Most often develops in the six decades of life and has a male predominance.
IgG4 accounts for less than 5% of the total IgG and healthy adults.
An isolated elevation of IgG4 levels is not diagnostic of IgG 4-related disease.
A combination of an elevated IgG4 level and typical imaging features such as diffuse or localized swelling of the pancreas is diagnostic as is autoimmune pancreatitis and retroperitoneal fibrosis.
Infiltrating plasmacytic IgG4 cells can lead to tissue destruction and fibrosis (Saeki T).
IgG4-related disease is often, but not always associated with elevated serum IgG4 concentrations.
IgG4 accounts for less than 5% of total IgG and is the lease abundant immunoglobulin of the IgG subclass.
Polyclonal hypergammaglobulinemia; complement levels decreased to 45% 36% may have nonspecific ANA and 20% nonspecific ANCA.
61% have CT scan abnormalities.
38% of patients who underwent PET scan showed hypermetabolic kidney lesions and found extrarenal lesions.
Glomerular lesions present in 16% with membranous nephropathy being predominant.
The sensitivity of an elevated IgG4 for diagnosing IgG4-related disease is approximately 70% and the specificity is 85%.
IgG1, IgG2, and IgG3 can vary in healthy people by a factor of more than 100, with the normal range 0.01-1.4 mg/mL, but IgG4 concentrations within individuals are stable.
Usually involves the pancreas but other organs including the liver, salivary glands, retroperitoneum and orbits can be involved.
Typically affects middle-age an older men, with a male to female ratio of 3 to 1 in contrast to other autoimmune diseases.
Many organs systems involved can lead to failure of vital organs such as pancreas, liver, biliary tree kidneys, or aorta.
Clinical manifestations are recognized in nearly every organ system.
Linked to autoimmune pancreatitis, with pancreatic tissue containing large numbers of IgG4-plasma cells.
Clinical manifestations include type 1 autoimmune pancreatitis,, cholangitis, chronic sclerosing sialadenitis, bilateral symmetrical, painless swelling of the lachrymal and salivary glands, chronic sclerosing dacryoadeniis, inflammatory orbital pseudotumors, retroperitoneal fibrosis, aortitis, fibrous variant of Hashimoto’s thyroiditis, sclerosing mastitis, pulmonary inflammatory pseudotumors, inflammatory pseudotumors of the kidney, hypophysitis, , pachymeningitis, prostatitis, cutaneous-pseudolymphoms, localized and generalized lymphadenopathy, and constructive pericarditis.
20 to 50% of all retroperitoneal fibrosis may be including IgG4 related disease.
IgG4-related disease is elevated in 7-10% of patients with pancreatic cancer, cholangiocarcinoma, primary sclerosing cholangitis, acute pancreatitis and healthy individuals.
IG4-Related disease may result from type 2 helper cell activation, in response to prolonged exposure to an unknown antigen.
Affects men slightly more than women, 3:1.
Average onset occurs at 50-70 years of age.
Nearly 80% of cases reported from Japan.
Elevated IgG4 levels reported in many cases of autoimmune pancreatitis, but the levels may be normal in cases of extrapancreatic disease.
Associated with elevation in the sedimentation rate, C-reactive protein level and peripheral eosinophilia may be seen.
Regional lymphadenopathy is common in autoimmune pancreatitis due to hyper IgG4.
Histological changes include storiform fibrosis, obliterative phlebitis, lymphoplasmacytic infiltration and eosinophilia.
Rarely elevated IgE the levels are present.
Diagnosis relies on clinical, lab, and histopathology , but no pathognomonic findings exist.
Diagnosis suspected in unexplained enlargement of 1 or more organs or tissues.
Histologic findings across affected organs are characterized by lymphoplasmacytic infiltration, fibrosis, and obliterative phlebitis.
Staining the pathological specimen for IgG4 facilitates calculation of the number of IgG4 cells.
4 lab studies suggest Dx: eosinophilia, hypergammaglobulinemia, elevated IgE levels, and hypocomplementemia.
Measuring IgG4 has limitations, precluding reliance on serum concentrations for diagnostic purposes.
Many patients have marked increased levels of IgG4.
Treatment:
No approved drug therapies for IGG for related disease at this time.
Corticosteroids are recommended for remission induction, and flare prevention.
Corticosteroids may improve the process, but control is not long-term when they are tapered or discontinued.
Rituximab is efficacious.
Agents targeting CD 19 may be effective.
Usually responds to corticosteroid therapy but bortezomib has been reported as effective (Khan M).
Inebilizumab, a humanized IgG1 kappa monoclonal antibody targeting CD 19 results in rapid, deep and durable B cell depletion:MITIGATE trial, it reduced the risk of flares of IgG4 related disease and increased the likelihood of flare free complete remission at one year, confirming the role of CD 19 targeted B-cell depletion as a treatment.
Patients may relapse when steroids are withdrawn and maintenance treatment may be needed.
Process encompasses 2 disorders: type I with IgG4-related disease and type 2 with substantial clinical overlap with type I but other distinctly pathological features.
IgG4- related disease described in virtually all organs including: biliary tree, salivary glands, periorbital issues, lungs, lymph nodes, kidneys, meninges, aorta, skin, thyroid, and pericardium.
IGG4 does not activate the classical complement pathway and may play only a limited role in immune activation.
Prominent treatment with corticosteroids is necessary when organ involvement is present.