 A disorder of the gut which is caused by the failure of the neural crest cells to migrate completely during fetal development of the intestine.
A disorder of the gut which is caused by the failure of the neural crest cells to migrate completely during fetal development of the intestine.
Congenital aganglionosis.
There is a lack of ganglia in the myenteric and sub mucosal plexuses of the gut.
A developmental disorder of the enteric nervous system and is the most common cause of intestinal obstruction in neonates and infants.
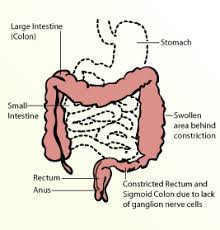
Hirschsprung’s disease is a congenital disorder of the colon in which nerve cells of the myenteric plexus (ganglion cells) in its walls are absent.
Hirschsprung’s disease is a form of functional low bowel obstruction.
Hirschsprung’s disease is due to failure of caudal migration of neuroblasts within developing bowel, with an absence of parasympathetic intrinsic ganglion cells in both Auerbach’s and Meissner’s plexuses.
The disease has more than 80% heritability, including significant associations in gene variance related to the enteric nervous system, as well as with monogenic and chromosomal syndromes.
About 18% of patients have multiple anomalies, some with specific syndromes, and approximately 12% have major chromosomal variants.
3-18% of siblings have the same disease.
Affected colon fails to relax, resulting in an obstruction.
Incidence of 15 cases per hundred thousand live births, and is characterized by high heritability and marked sex differences with a male to female ratio 4 to 1.
Associated with abdominal pain and most commonly diagnosed in the first year of life, but can present later in childhood.
Congenital condition characterized by partial or complete colonic obstruction associated with the absence of intramural ganglion cells.
Hirschsprung’s disease is highly heritable, and yet it is not obviously mendelian in its transmission through families.
Many patients with Hirschsprung’s disease have persistent gastrointestinal dysfunction, such as obstructive symptoms, fecal incontinence, or enterocolitis, despite removal of the aganglionic bowel.
Aganglionosis affects bowel segments of variable length, as a result of incomplete enteric neuronal colonization.
The condition may be classified as short, long, or total colonic aganglionosis.
Because of the aganglionosis, the lumen is tonically contracted, causing a functional obstruction.
The aganglionic portion of the colon is always located distally, but the length of the segment varies.
In most cases affects the short segment of the colon.
Rarely aganglionosis involves more than a segment of the colon, and in 5 percent of cases the entire colon is affected.
Occurs in approximately one in 5000 births.
Reflects a defect in the craniocaudal migration of neuroblasts originating from the neural crest that occurs during the first 12 weeks of gestation.
Appears on 18.6 per 100,000 live births.
Incidence of 1/5000 births
More common in males, and in Caucasians
Common among patients with Down syndrome.
Down Syndrome is comorbid in two percent of Hirschsprung’s cases.
Trisomy 21 is a risk factor for Hirschsprung’s associated enterocolitis.
May be caused by the interaction between two proteins encoded by two variant genes.
The RET proto-oncogene on chromosome 10 must interact with the gene EDNRB located on chromosome 13.
RET codes for proteins that assist cells of the neural crest , which later become ganglion cells, in their movement through the digestive tract during the development of the embryo, and the earlier the mutation occurs, the more severe the disorder becomes.
EDNRB codes for proteins that connect these nerve cells to the digestive tract.
RET is also with thyroid cancer and neuroblastoma and these disorders have also been observed in Hirschsprung’s patients with greater frequency than in the general population.
Associated with an absence of ganglionic cells and extending from the rectum proximately for a variable distance.
Only 6% of newborns with this disease pass meconium at 24 hours, and 37-54% to 48 hours.
Other symptoms include vomiting, explosive stools after examining the rectum, abdominal swelling ,gas and bloody diarrhea.
Enterocolitis is the most serious complication in children.
In infants and neonatal 12% have an initial presentation of Hirschsprung’s disease with enterocolitis.
Definitive diagnosis is made by suction biopsy of the distally narrowed bowel.
Diagnostic techniques involve anorectal manometry,barium enema, and rectal biopsy.
Alternative biopsies such as a deep mucosal for full thickness rectal biopsies can be done but had increased complication rate.
Specimen adequacy requires a mucosal biopsy measurement of 2-3 mm in diameter and 1 mm depth to make diagnosis.
Diagnosis of HD is confirmed by absence of ganglion cells on at the 75 consecutive 5 µm sections of tissue(Qualman SI et al).
Because tissue acquisition is not always adequate the diagnosis is supported by analysis of sub mucosal nerve fibers, histochemical analysis for acetylcholineesterase, and immunostaining for calretinin.
Mucosal biopsy specimens often reveal abnormal acetylcholinesterase positive neurites with in the muscularity mucosae.
Calretinin positive mucosal neuritis are absent in HD, while abnormal acetylcholinesterase positive neurites are present.
Submucosal hypertrophic nerves measuring more than 40 µm in diameter are characteristic of HD.
Treatment consists of surgical removal of the abnormal section of the colon, followed by reanastomosis.
The pull-through procedure repairs the colon by connecting the functioning portion of the bowel to the anus and is the standard treatment.
When aganglionosis extends proximal to the recto sigmoid, a combined transabdominal and perineal operation is performed.
Occasionally a colostomy above the aganglionic segment is performed and the child is for back month later for a definitive pull through surgery.
15% of children do not obtain full control, and laxatives or a high fiber diet and enema therapy may be necessary.
If the affected portion of the lower intestine is restricted to the lower portion of the rectum, other surgical procedures, such as the posterior rectal myectomy, may be done.
Mild Hirschsprung’s associated enterocolitis is treated with oral metronizadole and rectal dilatation to allow passage of stool.
Rectal irrigation wigwam saving to evacuate stool may be an important component of treatment for HD.
If rectal Irrigations or dilatations do not improve the patient, diversion with colostomy or ileostomy can be lifesaving.
Enterocolitis is to be cause of death with HD, but with earlier diagnosis and improved management of mortality rate has increased from 33% to approximately 1%.