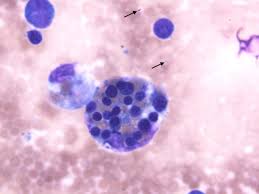
 Nonmalignant disease altering the mononuclear phagocytic system with histiocytes proliferation and prominent phagocytosis.
Nonmalignant disease altering the mononuclear phagocytic system with histiocytes proliferation and prominent phagocytosis.
A rare, frequently fatal syndrome of pathological immune activation, characterized by hyperactivity of lymphocytes and histiocytes, excessive cytokininerelease, and progressive multiorgan dysfunction.
The prevalence of HLH varies from country to country: in Sweden, the incidence is estimated to be approximately one case per 50,000 live births.
Familial HLH is an autosomal recessive syndrome and occurs most often in areas where consanguinity is common.
The median age of onset is 3 to 6 months.
The prevalence of secondary HLH is less well established and is largely underdiagnosed.
HLH is a rare syndrome caused by defects, either inherited or acquired, in cytotoxic T lymphocytes and/or natural killer cells leading to an uncontrolled inflammatory response by activated macrophages.
The underlying cause of familiar HLH is defective lymphocyte cytotoxicity.
Familial lymph HLH typically presents an infancy and occurs in approximately one in50,000 to 100,000 live births, with the rate higher in populations in which consanguinity is common.
Up to 25% of cases are currently classified as secondary HLH potentially linked to an underlined genetic immune disorder.
A French registry found the prevalence ranging from 3.8 to 15.4% per hundred thousand in children and from 1.3 to 5.4 per 100,000 in adults.
A life-threatening severe syndrome characterized by extensive inflammation that often leads to multiorgan failure and death, if not treated promptly.
Primary hemophagocytic lymphohistiocytosis is a rare syndrome characterized by immune dysregulation and hyperinflammation, that manifests in infancy and is associated with a high mortality.
Familial primary HLH, X-linked lymphoproliferative syndrome types, 1 and 2, and several albinism syndromes with immunodeficiency arising due to inherited defects and chains involved in cytotoxic granule-mediated apoptosis.
Familial HLH is caused by variants in four specific genes: PRF1, UNC 13 D, STX 11 and STXBP2 which encode proteins that are essential for normal functioning of natural killer cells (NK) and cytotoxic T Cells.
The underlying cellular defect in familial HLH is impaired lymphocyte cytotoxicity in NK cells and cytotoxic T cells.
Normally, these cells kill target cells, such as virus infected in cancer transformed cells by inducing programmedcell death in the immunologic synapse through activation of the performin-granzyme cell death pathway.
In familial HLH this process is deficient, resulting in insufficient production of performin or reduced secretion of performin containing granules from cytototoxic cells.
A disorder of pathological activation of the immune system and it often is fatal is not properly recognized and managed.
Primary HLH is typically genetic and often presents in children.
It is associated with defects in cytotoxic lymphocytes, particularly natural killer cells and cytotoxic T cells.
Primary HLH, also known as familial HLH (FHL), is often linked to specific genetic mutations that impair the cytotoxic function of natural killer (NK) cells and cytotoxic T lymphocytes.
Detective lymphocyte cytotoxicity causes an uncontrolled expansion of antigen specific effector T cells, sustained by the inability of CD8 positive T cells to deplete antigen presenting cells and defective down regulation of the immune response by cytotoxic cells.
In familial HLH initiation of the immune response is appropriate, but there is an inability to terminate it, and it leads to massive over activation of activated lymphoid inflammatory cells with markedly elevated levels of inflammatory cytokines, including interferon-gamma, IL-1 beta, IL-6, IL-10, IL-18, tumor necrosis factor and CXCL-9.
CXCL-9 is surrogate marker for interfere on-Gamma.
IL-gamma plays a pivotal role in both primary and secondary HLH.
Signs and symptoms of familial HLH reflect hypercytokinemia..
Typically, HLH presents in a septic like condition associated with cytopenias and have hepatosplenomegaly in a child, often in an infant with a febrile illness.
Thrombocytopenia, anemia and neutropenia are common.
HLH is often complicated by coagulopathy manifesting as thrombocytopenia, hypofibrinogenemia, prolonged clotting times, all associated with an elevated risk of bleeding and poor outcomes.
Hematophagocytosis is not always present and particularly not in the early phase of disease.
These include mutations in genes such as PRF1, UNC13D, STX11, and STXBP2, among others.
Specific genetic mutations linked to primary HLH include:
Familial HLH types 2-5 (PRF1, UNC13D, STX11, STXBP2) X-linked lymphoproliferative disease types 1 and 2 (SH2D1A, BIRC4) Griscelli’s syndrome type 2 (RAB27A) Chédiak–Higashi syndrome (LYST) Other defined mendelian disorders such as lysinuric protein intolerance (SLC7A7) and Wolman’s disease (LIPA).
Secondary HLH is acquired and more common in adults.
The overall estimated incidence of secondary HLH was 1 to 2 cases per million per year, but the incidence has significantly increased in the last two decades with an annual increase of 11%.
The overall rising incidence of HLH is attributed to increase the awareness, Improved diagnostic techniques and growing prevalence of associated comorbidities.
The majority of HL cases (70%) are an individuals age, 15 years and older, indicating a higher prevalence in adults than children.
It is often triggered by:
Infections: Viral (Epstein–Barr virus, cytomegalovirus), bacterial, parasitic, and fungal infections.
Malignancies: Hematologic cancers (e.g., lymphomas, leukemias) and solid tumors.
Autoimmune diseases: Conditions such as systemic-onset juvenile idiopathic arthritis, adult-onset Still’s disease, and systemic lupus erythematosus, often referred to as macrophage activation syndrome (MAS-HLH).
Other triggers include : Transplantation, iatrogenic immune activation or suppression, and other nonmendelian conditions.
X-linked lymphoproliferative diseases (SH2D1A, XIAP) and other genetic disorders like Griscelli syndrome type 2 (RAB27A) and Chédiak-Higashi syndrome (LYST) are notable causes.
Secondary HLH, on the other hand, can be triggered by a variety of conditions:Infections are a significant cause, particularly viral infections such as Epstein-Barr virus (EBV), cytomegalovirus (CMV), and other herpesviruses.
Bacterial, fungal, and parasitic infections can also precipitate HLH.
Secondary HLH or manifest as a critical illness with sepsis like manifestations that is unresponsive to sepsis direct therapy.
Malignancies, especially hematologic cancers like lymphomas and leukemias, are another major trigger, with lymphoid malignancies being the most common underlying cause in adults.
Various genetic lesions, background inflammation, underlying immunosuppression, and infectious triggers reach a threshold in which inflammation becomes uncontrolled and permanent HLH develops.
In secondary HLH, unlike the familial form, the circulating number of NK cells and cytotoxic cells are often reduced.
In adults with HLH, infections are the most common trigger (50%), followed by cancers (28%), and auto immune diseases it in 12%.
In children infections associated HLH is the most common form followed by auto immune diseases.
Malignancy associated HLH is much less common in children.
Autoimmune and autoinflammatory diseases, such as systemic lupus erythematosus and systemic-onset juvenile idiopathic arthritis, can also lead to HLH(macrophage activation syndrome (MAS).
HLH can also be associated with primary immunodeficiencies and inborn errors of metabolism.
Conditions such as lysinuric protein intolerance and lysosomal acid lipase deficiency (Wolman disease) are examples where metabolic disorders can lead to HLH.
Primary disease is a rare, autosomal recessive disorder of infancy either familial or sporadic.
Gene variants that cause primary HLH include PRF1, UNC1 3-D and STXBP2, which are the most common.
Classified into a primary-genetic, Mendelian form, and a secondary-acquired non-Mendelian form.
Neurologic symptoms occur less often in patients with secondary HLH than those with familial HLH affecting 10 to 25% of patients with secondary HLH.
Secondary disease associated with infection, malignancy, immunosuppression CAR-T cell therapy, and fat overload.
The primary form of HLH familial HLH typically affects children, mostly infants.
The secondary form of HLH is much more common in adulthood.
Secondary HLH is most commonly triggered by infections, cancer, and autoimmune disease.
HLH with an autoimmune trigger is referred to as macrophage activation syndrome.
It is largely under diagnosed and results in unnecessary deaths.
It can be diagnosed in adults due to underlying infection, auto immune disease, and malignant condition.
Life threatening syndrome with extreme inflammation due to an uncontrolled immune response.
Elevated bilirubin levels are a significant poor prognostic factor, particularly in pediatric populations.
There is an association with elevated liver function tests, and creatinine levels and adverse prognosis.
Respiratory failure is a common complication of HLH, often manifesting as acute respiratory distress syndrome.
Unrestrained and exaggerated inflammatory reactions.
Associated with ineffective immune response.
When hemophagocytic lymphohistiocytosis is associated with the setting of a rheumatological disease it is called macrophagevactivation syndrome.
Uncontrolled activation of the immune system lead to systemic inflammation, cytokine storm, and ultimately multiorgan failure.
Widespread infiltration of lymphocytes and histiocytes, leading to hemophagocytosis.
Characterized as primary, familial, or secondary, acquired.
Familial disease occurs in infants and young children with a family history of HLH, or an underlying genetic defect with dysfunctional cytotoxic T lymphocyte cells, and natural killer (NK) cells.
Acquired HLH occurs in adults due to infectious, malignant or autoimmune disorders.
The most common conditions associated with HLH are malignancy, at about 50%, infections at 30%, auto immune disorders 10%, and idiopathic disease at 10%.
Occurs in an estimated 12-17% of cases of adult onset Still disease.
Lymphoproliferative disorders of the B and T cell origin are the most common causes of secondary HLH accounting for 68% of cases over the age of sixty years, followed by infections at 26%, and autoimmune disorders at 6%.
Clinical criteria required for the diagnosis involves five of the eight following criteria:
Fever
Splenomegaly
Cytopenia of two or more cell lines
Hypertriglyceridemia and or hypofibrinogenemia
Hematophagocytosis in bone marrow/lymph nodes/spleen
Low or absent NK-cell activity
Ferritin greater than 500 mcg/L
Soluble CD25 (IL 2 receptor) activity of greater 2400 units per mL.
Interferon gamma inhibits lipoprotein lipase which increases triglyceride levels.
Secretion of plasminogen activator by activated macrophages causes increased plasmin levels, hyperfibrinolysis, and decreased fibrinogen.
Ferritin production is increased by the macrophage enzyme heme oxygenase enzyme.
Elevated ferritin levels, is a relatively consistent findings in HLH adults.
Interleukins 1 and 6 are increased and produce fever and soluble IL-2 receptor increases in response to activation of Th1 cells.
Organ infiltration of the liver and spleen account for hepatosplenomegaly and neurological symptoms are a result of activation of lymphocytes in histiocytes.
Neurologic symptoms may include visual problems, seizures, irritability, altered mental status and may occur in up to 37% of patients.
Clinical features include fever, multi organ dysfunction, CNS issues.
HLH can be primary owing to genetic defects involving cytotoxic T cells and natural killer cells or secondary induced by viral infections, and autoimmune disorders and is usually associated with liver injury.
Pleocytosis and hematophagocytosis typically present in CSF fluid.
5-year survival in children is about 54%.
In an adult study of 250 adult patients the median age was 49 years, median survival 2.1 months for the entire group (Parikh, SA et al).
In the above study median survival is 1.4 months with malignant associated disease.
In the presence of malignancy or hypoalbuminemia the prognosis is poor.
Treatment of adult HLH is based on correcting the underlying precipitating factor and the associated hyper inflammatory response.
Patients with infection associated HLH can be treated adequately for the underlying infection.
The addition of immunosuppressive and corticosteroids. cyclosporine or intervenous immunoglobulins are consider in patients with severe hyperinflammatory responses.
In EBV associated HLH etoposide based treatment has shown improvement in outcomes, have the addition of rituximab.
Emapalumab (Gamifant) is the first treatment specifically approved for primary hemophagocytic lymphohistiocytosis.
A combination of liposomal doxorubicin, etoposide , and high-dose methylprednisone had an overall response rate of 76%: median survival only seven months.
The most utilized treatment protocols for primary HLHR based on the use of etoposide, dexamethasone and cyclosporine.
Pre-treatment of hemopoietic stem cell transplant with the above protocol has improved survival levels.
In patients with secondary HLH treatment is adapted to the underlying condition and severity of the HLH with treatment regimen similar to that for primary HLH and include additional agents of interleukin-1 and JAK inhibitors as well as intravenous gamma globulin.