 A group of heterogeneous diseases, with more than 20 forms with variable clinical severity.
A group of heterogeneous diseases, with more than 20 forms with variable clinical severity.
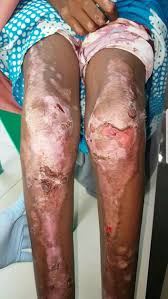
Characterized by loss of skin integrity leading to blisters and skin erosions.
Affects approximately 1 to 3 persons per million in the United States.
Recessive dystrophic epidermolysis bullosa is one of the most severe forms, and it is caused by a loss of function mutation in the collagen type VII gene (COL7A1).
A rare genetic blistering disease caused by mutations in COL7A1 gene encoding type VII collagen resulting in absent or dysfunctional anchoring fibers and which disrupt adhesion of the epididymis to the dermis.
Caused by mutations in genes that encode collagens and laminins, components of the extracellular matrix that are essential for maintaining the structural integrity of skin.
It can be inherited in an autosomal dominant or recessive manner.
There is a loss of anchoring, fibrils and structural integrity in the superficial dermis due to mutations in the gene encoding type VII collagen (COL7A 1).
The loss of function mutation in collagen type VII (C7) gene markedly diminishes the expression of C7, a collagen at the epidermal-epidermal junction.
C7 is the primary component of the anchoring fibrils that tether the epidermal basement membrane to the dermal matrix.
The C7 proteins is the major component of anchoring fibrils responsible for the binding of the epidermis and dermis, and in dystrophic epidermolysis bullosa, the attachment is disrupted, leading to skin fragility and wounds involving the entire body surface area, as well as extra cutaneous areas of the oral, ocular, Gastrointestinal, and genitourinary systems.
With impaired C7 expression skin collagen fibrils do not form properly and normal epidermal-dermal adherence beneath the lamina densa of the basement membrane is lost.
Patients with recessive dystrophic epidermolysis bullosa experience painful skin erosions and blister formation on mucosal membranes and skin, resulting in esophageal strictures, severe scar formation, local and systemic infections, joint contractions, finger and toe fusions, and aggressive squamous cell carcinomas of the skin.
There is extreme mechanical fragility of the skin, which tears with the slightest touch.
Minor trauma from every day activities causes, repeated, blisters and scars of epithelial tissues, leading to esophageal and genitourinary structures, mitten deformities from adhesion of the digits, malnutrition, anemia, and infection.
Itching and pain can impair the quality of life.
Squamous cell cancers, which develop from chronic wounds represent the leading cause of death in adults with dystrophic epidermolysis bullosa.
Repeated blistering and fibrosis can lead to squamous cell carcinoma, life-threatening infections, and limb deformities.
Recessive dystrophic epidermolysis bullosa had a median survival of 55 to 65 years (Fine JD).
A variant of recessive dystrophic epidermolysis bullosa the Hallopeau-Siemens disease is associated with severe mucosal and cutaneous blistering apparent at birth and associated with the median survival of approximately 30 years.
Management of patients with recessive dystrophic epidermolysis bullosa is primarily palliative treatment of wounds.
In a study of children with recessive dystrophic epidermolysis bullosa treated with allogeneic bone marrow transplantation indicated that increased C7 deposition, was present with improved healing of wounds and reduction in blister formation and sustained presence of donor cells found in the skin with increased C7 deposition (Wagner JE).
In the above study, allogeneic bone marrow was demonstrated to partially correct the C7 deficiency and improve skin and mucosal healing in recessive dystrophic epidermolysis bullosa.
Bone marrow transplantation in the small group was associated with a 30% mortality.
Complete wound dealing at three and six months in patients with dystrophic epidermolysis bullosa was is more likely with topical administration of Beremagene geperpavec (B-VEC) than with placebo.
Beremagene geperpavec (B-VEC) is a topical herpes simplex virus type 1 based gene therapy designed to restore C7 protein by delivering COL7A1: it is associated with a significantly higher percentage of wounds with complete healing at three and six months, then application of placebo.
Patients with dystrophic Epidermolysis bullosa may have ocular findings, including corneal erosion, scarring, blistering, and erosion that may give way to chronic scarring and progressive vision loss: B-VEC is potentially beneficial