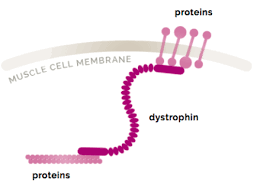
 Dystrophin play a major role in the function of cardiac myocytes and skeletal myofibers as part of the dystrophy – glycoprotein complex that anchors myofilaments to the extracellular matrix and prevents stress induced damage to the sarcolemma membrane.
Dystrophin play a major role in the function of cardiac myocytes and skeletal myofibers as part of the dystrophy – glycoprotein complex that anchors myofilaments to the extracellular matrix and prevents stress induced damage to the sarcolemma membrane.
DMD gene is located on the short (p) arm of the X chromosome between positions 21.2 and 21.1
Dystrophin is a rod-shaped cytoplasmic protein, and a vital part of a protein complex that connects the cytoskeleton of a muscle fiber to the surrounding extracellular matrix through the cell membrane.
Many muscle proteins, such as α-dystrobrevin, syncoilin, synemin, sarcoglycan, dystroglycan, and sarcospan, colocalize with dystrophin at the protein complex.
It has a molecular weight of 427 kDa.
Dystrophin is coded for by the DMD gene – the largest known human gene, covering 2.4 megabases making up 0.08% of the human genome, at locus Xp21.
Spontaneous or inherited mutations in the dystrophin gene can cause different forms of muscular dystrophy, a disease characterized by progressive muscular wasting.
The most common of these disorders caused by genetic defects in dystrophin is Duchenne muscular dystrophy.
It is a protein located between the sarcolemma and the outermost layer of myofilaments in the muscle fiber.
It is a cohesive protein, linking actin filaments to other support proteins that reside on the inside surface of each muscle fiber’s plasma membrane.
These support proteins on the inside surface of the sarcolemma in turn links to two other consecutive proteins for a total of three linking proteins.
Dystrophin supports muscle fiber strength.
The absence of dystrophin reduces muscle stiffness, increases sarcolemmal deformability, and compromises the mechanical stability.
Dystrophin deficiency is one of the main causes of the general class of myopathies, referred to as muscular dystrophy.
The deletions of one or several exons of the dystrophin DMD gene can result in the disease known as Becker’s muscular dystrophy which has a milder phenotype and longer survival.