Bardet–Biedl syndrome (BBS) is a ciliopathic genetic disorder that produces many effects and affects many body systems.
It is characterized by rod/cone dystrophy, polydactyly, central obesity, hypogonadism, and kidney dysfunction in some cases.
Slower mental processing maybe present.
Biedl-Bardet Syndrome is an autosomal recessive process; but epigenetic phennomena also cause some of the variation seen in BBS.
It has variable expressivity and a wide range of clinical variability observed both within and between families.
Its most common clinical features:
are rod–cone dystrophy, with childhood-onset night-blindness followed by increasing visual loss;
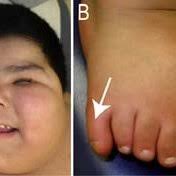
postaxial polydactyly;
truncal obesity that manifests during infancy and remains throughout adulthood;
varying degrees of learning disabilities;
male hypogenitalism and complex female genitourinary malformations; and renal dysfunction, a major cause of morbidity and mortality.
A wide range of secondaryare sometimes associated with BBS.
Strabismus, cataracts, astigmatism, pigmentary retinopathy, poor visual acuity, low vision, and/or blindness caused by an impaired photoreceptor transport mechanism in the retina.
Brachydactyly, syndactyly of both the hands and feet is common, as is partial syndactyly.
Polyuria/polydipsia with nephrogenic diabetes insipidus
Ataxia/poor coordination/imbalance
Mild hypertonia, especially of the lower limbs.
Diabetes mellitus
Hepatic involvement
Anosmia
Auditory deficiencies
Hirschsprung disease and subsequent bowel obstruction.
Hypertrophy of interventricular septum and left ventricle and dilated cardiomyopathy.
Hypogonadism, kidney failure, urogenital sinuses, ectopic urethra, uterus duplex, septate vagina, and hypoplasia of the uterus, ovaries, and fallopian tubes.
Speech disorder/delay
Developmental delay-gross motor skills.
BBS is caused by defects in the cellular ciliary structure, and is is a ciliopathy.
Ciliopathies include: primary ciliary dyskinesia, polycystic kidney and liver disease, nephronophthisis, Alström syndrome, Meckel–Gruber syndrome and some forms of retinal degeneration.
The gene products encoded by these BBS genes, called BBS proteins, are located in the basal body and cilia of the cell.
BBS proteins are involved in a process called intraflagellar transport (IFT), a bi-directional transportation activity within the cilia along the long axis of the ciliary shaft that is essential for ciliogenesis and the maintenance of cilia.
Mutated BBS genes affect normal cilia function, which, in turn, causes BBS.
That photoreceptor cells are nourished by the IFT of retinal cilia now offers a potential explanation for the retinal dystrophy common in BBS patients after their early years of life.
Diagnosis:
The diagnosis of BBS is established by clinical findings and family history.
Molecular genetic testing can be used to confirm the diagnosis.
There is currently no specific treatment.