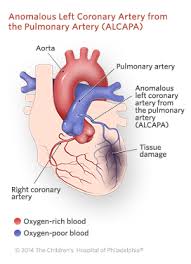
 Anomalous left coronary artery from the pulmonary artery (ALCAPA, Bland-White-Garland syndrome) is a rare congenital anomaly occurring in approximately 1 in 300,000 live-born children.
Anomalous left coronary artery from the pulmonary artery (ALCAPA, Bland-White-Garland syndrome) is a rare congenital anomaly occurring in approximately 1 in 300,000 live-born children.
The diagnosis comprises between 0.24 and 0.46% of all cases of congenital heart disease.
The anomalous left coronary artery (LCA) usually arises from the pulmonary artery instead of the aortic sinus.
In fetal life, the high pressure in the pulmonic artery and the fetal shunts enable oxygen-rich blood to flow in the LCA.
By the time of birth, the pressure will decrease in the pulmonic artery and the child will have a postnatal circulation.
The myocardium which is supplied by the LCA, will therefore be dependent on collateral blood flow from the other coronary arteries, mainly the RCA.
Because the pressure in RCA exceeds the pressure in LCA a collateral circulation will increase.
This situation ultimately can lead to blood flowing from the RCA into the LCA retrograde and into the pulmonary artery, thus forming a left-to-right shunt.
The development of symptoms in ALCAPA depends heavily on the amount of collaterals development.
When only few collaterals are present, the myocardium will not get enough oxygen and will become ischemic.
The symptoms in an infant with ALCAPA include signs of heart failure such as dyspnea and
Sometimes the first sign of ischemia can be crying during feeding, sweating, failure to thrive and irritability.
Approximately 90% of patients die within the first year if left untreated.
Patients with a significant collateral circulation can live to adulthood in rare cases, but their they develop chronic ischemia, having a risk for sudden cardiac arrest, heart failure or malignant arrhythmia.
Historically ALCAPA was diagnosed with conventional angiography.
Today echocardiography can provide direct visualization of the anomalous coronary artery and other associated structural abnormalities, and it can also assess myocardial function.
The use of pulse and color-flow doppler can sometimes visualize reversal flow in the pulmonic artery.
Other non-invasive methods used are computed tomography (CT) andmagnetic resonance imaging (MRI) enable a direct visualisation of the arteries as well as the myocardial viability.
Surgery is indicated in all patients with ALCAPA independent of symptoms.
The reconnection of the anomalous left coronary artery, to the aortic root, is crucial to the perfusion of the myocardium dependent on that vessel.
Establishment of a dual coronary system is the preferred method and if possible reimplantation of the artery is the approach of choice.
Even if surgery is carried in adulthood, reestablishment of a two-coronary system can make malignant arrhythmia disappear.
No difference in long-term mortality or left ventricle function has been shown between the different techniques to re-establish a two-coronary system.
The development of surgical techniques and restoring of two-artery circulation has dramatically increased survival.