 Mature peripheral T cell lymphoma comprising 15-20% of all peripheral T cell lymphomas and 1-2% of non-Hodgkin’s lymphomas.
Mature peripheral T cell lymphoma comprising 15-20% of all peripheral T cell lymphomas and 1-2% of non-Hodgkin’s lymphomas.
Linked to many viral infections including Epstein-Barr virus, Cytomegalovirus and herpes 6 virus.
Arises from CD4 T follicular helper cells.
Angioimmunoblastic T cell lymphoma (AITL) , the second most common form of mature T cell lymphoma, is widely considered aggressive and can be complicated by Epstein‐Barr virus (EBV) infection and the co‐development of EBV‐driven diffuse large B‐cell lymphoma.
Epstein-Barr virus infections are present in 66 to 91% of patients with AITL.
With 80% of patients have nodal or extranodal disease at the time of diagnosis.
Patients typically present with an acute illness with systemic manifestations of fever, weight, loss, and/or night sweats, and generalized lymphadenopathy.
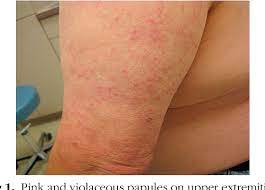
skin manifestations occur in up to 50% of patients and are usually nonspecific maculopapular eruptions mimicking drug/viral exanthemas and plaque-like nodular lesions, that are more likely to be recognized as lymphoma.
Median age at diagnosis is 65 years, occurring mainly in older patients.
There’s a slight male predominance.
Affects mainly elderly men with a median age of 60 years.
Moderate to highly aggressive course.
Rarely can spontaneously remit.
Most present with generalized lymphadenopathy, hepatosplenomegaly, skin rash, fever and weight loss.
Patients may present with pleural effusions and ascites or peripheral edema.
Commonly associated with polyclonal hypergammaglobulinemia, positive Coomb’s test and positive ANA.
Immune system mediated anbnormalties are common in AITL, and include elevated ESR, hemolytic anemia, cryo-globulins, and polyclonal hypergammaglobulinemia.
Rash can be confused with vasculitis demonstrates perivascular dermal infiltrate with eosinophils, plasma cells, and lymphoid cells.
Genetic mutations are most common in Angioimmunoblastic T cell lymphomas which is the second most common form of mature T cell lymphomas, carries a very poor prognosis, and is considered relatively chemotherapy‐resistant.
The disease is considered highly aggressive and presents with diffuse lymphadenopathy and often cutaneous disease.
The cell of origin is the follicular Th cell normally present in the germinal center of lymph nodes and is defined by expression of CD4, CD10, Bcl‐6, PD‐1, and CXCL13.
For diagnosis T cell rearrangement studies needed because B and T cells are usually present in diagnostic biopsies.
IDH2 mutation has been identified in 16 approximately 20% of patients with Angioimmunoblastic T cell lymphomas.
Lymph node histology reveals partial effacement of nodal architecture with a polymorphous infiltrate.
Arborization and proliferation of endothelial venules along with follicular dendritic cells.
Derived from CD2, CD3, CD4, CD5 and CD7 positive helper T cells.
The presence of helper T cells and eosinophils and hypergammaglobulinemia suggest chronic absorption of a CD4 stimulating antigen initiates the disease.
Aggressive lesion with poor prognosis.
The five-year survival is 32 to 41%.
Cyclophosphamide, doxorubicin, vincristine, bleomycin, procarbazine, prednisilone, ifosphamide, methotrexate and etoposide (COP-BLAM/ IMVP-6) (Siegert), with a 64% complete response rate and median survival of 15 months.
CHOP-like treatment associated with a 60% complete response and 36% 5-year survival.
Long term survival has been reported with autologous stem cell transplant when patients are in first remission or in the presence of chemosensitive recurrent disease.
Five-year survival after anthracycline based chemotherapy is 26% for peripheral T cell lymphomas and 32% for angioimmunoblastic T-cell lymphomas.