 Accounts for 5-15% of cases of acute myeloblastic leukemia.
Accounts for 5-15% of cases of acute myeloblastic leukemia.
The incidence of APL was 0.33 cases per 100,000 population per year.
The disease incidence increases significantly with age up to 79 years.
Incidence varies significantly by age, sex, race/ethnicity, and diagnosis period.
The increasing incidence over time could be related to multiple factors, including heightened awareness, diagnostic accuracy, better reporting and capturing of new cases, and obesity, which has been associated with APL.
Disease incidence is lower in women compared with men, whereas it was significantly higher among Hispanic versus non-Hispanic patients.
Hispanic patients constituted 21.5% of patients and had a higher disease incidence, similar to reports of increased incidence of APL and acute lymphoblastic leukemia in this population.
It represents a hematologic emergency because of the risk of bleeding from hyperfibrinolysis, disseminated intravascular coagulation, and thrombocytopenia.
M3 subtype of acute myelogenous leukemia in the French-American-British classification system.
Characterized characterized by accumulation immature white blood cells, promyelocytes in the bone marrow.
Associated with a severe hemorrhagic diathesis that leads to disseminated intravascular coagulation (DIC) and hyperfibrinolysis.
In 40% of untreated patients, pulmonary and cerebral hemorrhages can occur.
With treatment it takes 5-8 days for coagulopathy to improve.
Acute promyelocytic leukemia (APL) is a is a subtype of acute leukemia characterized by abnormal proliferation of promyelocytes, life-threatening coagulopathy, and the chromosome translocation t(15;17)(q22;q11-12).
APL has been transformed from a highly fatal disease to a highly curable one.
More than 95% of cases are characterized by a balanced translocation between chromosome 17q21 and chromosome 15q22.
The translocation between chromosome 17q21 and chromosome 15q22 leads to an abnormal fusion protein called PML-RARA.
This translocation can be detected by karyotyping or fluorescence in situ hybridization (FISH) studies, and by polymerase chain reaction (PCR) techniques.
The RT-PCR assay can establish the diagnosis of APL when cytogenetics and fluorescence in situ hybridization (FISH) fail.
The peripheral blood RT-PCR needs to be monitored every 3 months for the first 2 years: Then, the assay can be performed every 3-6 months for the next 3 years.
The retinoic acid alpha receptor gene (RARA) is encoded by the long arm of chromosome 17.
The retinoic acid alpha receptor gene is mainly expressed in hematopoietic cells and has an important role in regulating gene expression.
In the presence of retinoic acid, RARA is activated and terminal differentiation of promyelocytes occurs.
The promyelocytic gene (PML) is encoded by the long arm of chromosome 15.
The promyelocytic gene is involved in apoptosis and tumor suppression.
There are three possible isoforms caused by PML-RARA translocations.
The breakpoint in chromosome 17 is consistently found in intron 2.
The breakpoint varies in chromosome 15: The three breakpoints on the PML gene can occur at intron 3 (L form), intron 6 (S form), and exon 6 (V form).
The S form breakpoint is associated with a shorter remission duration and overall survival compared with the L form.
The fusion gene product causes the retinoic acid receptor to bind more tightly to the nuclear co-repressor factor, so that the gene cannot be activated with physiologic doses of retinoic acid.
In about 5% of cases, rearrangements of chromosome 17q21 with other gene partners occur.
About 40% of APL cases express additional chromosomal abnormalities, trisomy 8 and isochromosome 17.
The age-adjusted annual incidence is 0.23 per 100,000 persons.
Approximately 30,800 cases of acute leukemia are diagnosed yearly in the US, and about 1000 of those are APL.
The incidence in males and females is equal.
The median age at diagnosis is 40-44 years which is younger than most patients with AML
Its incidence is higher in patients originating in Latin America.
Blacks have lower lifetime incidence rates than non-Hispanic whites, Hispanics, and Asians.
Laboratory evaluation of acute promyelocytic leukemia (APL) should include:
Complete blood cell count with differential
Peripheral blood smear review
Comprehensive metabolic profile for baseline renal and liver function tests
Electrolyte levels
Prothrombin time (PT) and activated partial thromboplastin time (aPTT)
Fibrinogen assay
Some authorities suggest lumbar puncture at diagnosis in high-risk patients who present with a very high WBC count, as the CNS may serve as a sanctuary site warranting intrathecal therapy.
Prior to lumbar puncture any coagulopathy should be corrected first.
Flow cytometry of CSF should be analyzed for abnormal clonal cells.
A bone marrow biopsy with aspirate should be performed and sent for flow cytometry and cytogenetics.
Fluorescent in situ hybridization (FISH) for the translocation or reverse transcription polymerase chain reaction (RT-PCR) for the PML-RAR alpha transcript is performed.
Acute promyelocytic leukemia (APL) is myeloperoxidase positive and CD33 positive, human leukocyte antigen (HLA)-DR negative.
Cytogenetically has translocation of genetic material between 2 chromosomes (t[15:17] specifically a fusion of the promyelocytic leukemia gene on chromosome 15 to the retinoic acid receptor alpha (RARA) gene on chromosome 17.
Secondary to a block in the normal maturation process of myeloid progenitors at the promyelocyte stage.
The PML gene encodes a protein that suppresses tumors and interacts with cell proliferation and apoptosis.
In APL alteration of the PML gene prohibits these above functions and the protein produced by the fused gene PML-RARA causes abnormal cell proliferation and blocks differentiation of WBCs at the promyelocyte stage.
Manifests by cytopenias and coagulopathy.
Physical examination findings include: pallor, petechiae, ecchymoses, gum bleeding,and neurologic deficits or headaches if CNS involvement is present.
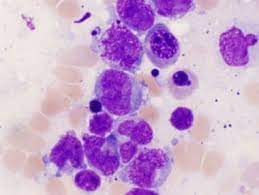
Histologic findings:
Hypergranular
Microgranular
Hyperbasophilic
The hypergranular subtype has frequent Auer rods, which are clumps of granular material containing lysosomes, peroxidase, lysosomal enzymes, and large crystalline inclusions.
While Auer rods can be seen in other types of AML, but they are usually seen in APL.
In APL the nucleus is folded or bilobed, and the cytoplasm contains prominent azurophilic granules.
The bone marrow usually appears hypercellular.
APL cells stain intensely for Sudan black and myeloperoxidase.
APL cells are periodic acidï Schiff (PAS) and HLA-DR statin negative .
The microgranular variant of APL has a folded nucleus, with cytoplasm that is fine, dusky granules and only rare Auer rods.
The microgranular variant of APL is seen in 25% of cases of APL.
A hyperbasophilic subtypes APL shows an increased nucleocytoplasmic ratio and strongly basophilic cytoplasm with blebs.
A hyperbasophilic subtype APL has few granules and no Auer rods.
Characterized with presence of granules and multiple Auer rods.
Most patients present with pancytopenia.
About 10-30% of patients present with leukocytosis.
Its coagulopathy has been described as disseminated intravascular coagulation (DIC) with associated hyperfibrinolysis.
ATRA helps to rapidly control the disseminated intravascular coagulation.
Treatment of disseminated intravascular coagulation (DIC) should also include platelet transfusions to maintain the platelet count at about 20,000/?L and cryoprecipitate to maintain a fibrinogen level at least above 100-150 mg/dL.
Majority of patients present with a significant degree of coagulopathy.
Because of pancytopenia patients commonly present with bruising, epistaxis, hematuria, petechiae, fever, fatigue, anorexia, pallor, weight-loss, and joint pain.
Symptoms: Fatigue, weakness, and dyspnea related to anemia, easy bruising or bleeding caused by thrombocytopenia or coagulopathy, fever and infection related to leukopenia.
Patients are at high risk of death from internal hemorrhage within a few hours after presentation and as such the diagnosis is considered a medical emergency.
The incidence of early death is high.
ATRA transformed this rapidly and invariably fatal form of leukemia and into a highly curable disease entity with long-term survival exceeding 80% in clinical trials.
Early deaths occurring within the first 30 days of diagnosis occurs primarily due to hemorrhage from disease related DIC and non-hemorrhagic causes such as infection and ATRA induced differentiation syndrome.
Early death rates range from 2 to 6% in clinical trials and from 17 to 37% in population based registries.
Measures to reduce early deaths include supportive care, aggressive weight management, steroids at induction and dose reduction of ATRA for adults 65 years and older.
There is a high propensity of patients with obesity.
Additional risk factors for early death include:
Increased serum creatinine level
Older age
Male sex
Elevated fibrinogen level
29% of patients die within 30 days of their diagnosis, but in 35% of those early deaths, the patient never received all-trans retinoic acid (ATRA) therapy.
Once the diagnosis is suspected targeted therapy and supportive transfusion therapy should be initiated immediately.
Coagulopathy is responsible for more than 60% of early deaths in APL.
Failure to enter complete remission is almost exclusively due to early deaths from hemorrhage, or APL differentiation syndrome
Treatment of disseminated intravascular coagulation (DIC) should also include platelet transfusions to maintain the platelet count at about 20,000/?L and cryoprecipitate to maintain a fibrinogen level at least above 100-150 mg/dL.
In patients who have central nervous system (CNS) involvement or who are at higher risk for CNS relapse, intrathecal chemotherapy is usually given.
The elderly have higher death rates in complete remission.
Intracranial hemorrhage accounts for 65-80% of presenting bleeding complications in APL.
Following intracranial hemorrhage bleeding complications include gastrointestinal hemorrhage and diffuse intra-alveoli hemorrhage of the lung.
Induction failure causes include: hemorrhage, infection, and APL differentiation syndrome.
About 25-50% of patients who receive ATRA develop differentiation syndrome, this usually occurs within the first 21 days of treatment.
A rapid increase in WBC count may follow ATRA use, and these patients should be promptly treated with chemotherapy to avoid clinical hyperleukocytosis.
Differentiation syndrome is characterized by the following:
Fever
Hypotension
Weight gain
Respiratory distress
Serositis with pleural or pericardial effusions
Hypoxemia
Radiologic infiltrates
Acute kidney injury
Hepatic dysfunction
With differentiation syndrome patients should be treated with intravenous steroids to prevent treatment-related mortality.
For high risk patients (WBC >10,000/?L), prophylaxis against differentiation syndrome is with corticosteroids with the dose tapered over several days.
With the differentiation syndrome the initiation of dexamethasone therapy at the first sign of symptoms of respiratory compromise should take place.
Temporary discontinuation of ATRA therapy should be considered until hypoxia resolves.
Hyperleukocytosis is common in differentiation syndrome, but WBC counts may also be normal.
Infection is seen more frequently in men older than 60 years and in patients who have fever at presentation.
Thrombosis occurs in up to one quarter of patients with APL and approximately 1/3 of these thrombotic complications occur following induction treatment.
Almost all patients have signs of DIC, have increased prothrombin time, increased partial thromboplastin time and thrombin time.
Fibrinogen levels and platelet counts are decreased and fibrin degradation products are increased.
Fibrinolytic system abnormalities are present with increased levels of tissue plasminogen activator (tPA), urokinase plasminogen activator, urokinase plasminogen activated receptor, and fibrinolytic receptor annexin A2.
It is associated with low levels of plasminogen, alpha2-plasmin inhibitor, and plasminogen activator inhibitor 1 found in fibrinolytic states.
There is increased expression of annexin II, a receptor for plasminogen and plasminogen-activating factor, that leads to overproduction of plasmin and fibrinolysis.
The coagulopathy is a medical emergency.
Patients have a clinical picture of both pro-fibrinolytic and anti-fibrinolytic factors varying from one patient to another.
Factors that increase risk of bleeding include high white blood count, thrombocytopenia, low fibrinogen levels, and the presence of infection.
Anti-fibrinolytic factors are increased including plasminogen activator inhibitor (PAI)-1 and plasminogen activator inhibitor-2.
Therapy-related myeloid neoplasms are a rare but severe complication.
Extramedullary disease or granulocytic sarcoma is present in 2.5-9.1% of cases.
Extramedullary disease is a rare initial presentation, but is largely seen in the setting of relapsed disease.
Extramedullary disease is usually seen in the CNS, and carries a poor prognosis.
Prognosis is associated with white blood cell count: low risk (WBC ?10,000/?L) or high risk (WBC >10,000/?L).
More common in Hispanics.
Majority of patients have a translocation that fuses the promyelocytic leukemia gene on chromosome 15 with a gene encoding a retinoic acid receptor on chromosome 17((t(15;17)(q22;q21).
A neutropenic diet should be ordered for leukopenic or neutropenic patients.
No fresh fruits or flowers should be allowed in the patient’s room.
The most important complications of acute promyelocytic leukemia (APL): are bleeding diathesis secondary to underlying coagulopathy (5%), infection (2.3%), and differentiation syndrome (1.4%).
Gene translocation creates the PMR-RARA and RARA-PML fusion genes resulting in the PML-RAR? fusion protein that determines the phenotype of acute promyelocytic leukemia and the response to all-trans-retinoic acid and arsenic trioxide treatment.
PML-RARA determines the phenotype of the disease with block of differentiation by repressing of genes implicated in myelopoiesis and which is overcome by retinoic acid.
PML-RARalpha can be detected by cytogenetic analysis, fluorescence in situ hybridization (FISH), or polymerase chain reaction) PCR) and molecular testing for the PML-RARalpha fusion transcripts.
Testing by immunohistochemistry, cytochemistry, and molecular genetic analysis are needed to diagnose APL.
Diagnosis requires APL peripheral morphology and one of the following:
t(15:17) by cytogenetics, or Promelocytic leukemia (PML)/retinoic acid receptor alpha (RARA) by molecular testing.
Patients are classified as low risk (white blood cell [WBC] count ?10,000/?L) or high risk (WBC >10,000/?L).
In the presence of neurologic signs or symptoms, brain imaging studies should be performed to detect meningeal disease, chloromas, or central nervous system (CNS) bleeding.
With conventional chemotherapy utilizing anthracyclines with or without cytosine results in complete remission rates of 70-80% and 40% cure rate.
With modern therapy cure rates exceed 70%, and should be 100%.
With targeted therapies, such as all-trans retinoic acid (ATRA), arsenic trioxide (ATO), and other agents, have current cure rates exceeding 90%.
Long-term survival rates up to 90% with treatment.
The multidrug resistance inhibitor P-glycoprotein, which extrudes chemotherapy from the internal part of the cell, seems to be lowest in patients with this type of leukemia and accounts probably for the increased sensitivity to anthracycline drugs.
Management: immediate institution of all-trans-retinoic acid (ATRA) and/or arsenic trioxide (ATO) treatment and supportive therapy is indicated.
ATRA may cause DNA fragmentation and damage or degrade the fusion protein PML-RAR alpha.
Used in APL patients who have the presence of the t(15;17) translocation or PML/RAR-alpha gene expression.
It is indicated also for induction of remission and consolidation in patients with APL who are refractory/relapsed to retinoid and anthracycline chemotherapy and in combination with tretinoin for treatment of adults with newly diagnosed low risk APL.
Side effects of ATRA include the following:
Headache
Nasal stuffiness
Dry, red skin
Pseudotumor cerebri
ATRA alone cannot eradicate the malignant clone, and to achieve a complete hematologic and molecular remission requires the addition of arsenic trioxide (ATO) or chemotherapy.
Cytogenetic variants of APL, especially cases with the PLZF-RARA mutation ,may manifest ATRA resistance.
Resistance may also develop as a secondary event in PML-RARA APL.
Treatment has three phases: induction, consolidation, and maintenance.
All-trans-retinoinic acid is an important agent in all three phases of APL treatment , as it can lead to terminal differentiation of malignant promyelocytes into mature neutrophils.
Induction therapy in patients with acute promyelocytic leukemia (APL) who are at low or intermediate risk recommendation is all-trans-retinoic acid (ATRA), 45 mg/m2 in divided doses daily until clinical remission, plus arsenic trioxide (ATO), 0.15 mg/kg IV daily until bone marrow remission.
The NCCN recommends in low- and intermediate-risk patients, to continue induction therapy until count recovery occurs, then proceeding to consolidation therapy.
For consolidation therapy in acute promyelocytic leukemia (APL), it is recommended basing the choice of regimen on the agents used for induction therapy.
The role of maintenance therapy remains uncertain, especially for patients with low-risk APL who achieve molecular remission at the end of consolidation treatment.
In high-risk patients receiving ATRA/ATO, induction is continued until count recovery and bone marrow remission are demonstrated.
Alternative regimens :
ATRA plus daunorubicin
ATRA plus idarubicin
In the French-Belgian-Swiss study revealed that the early addition of chemotherapy lead to significantly better survival, and the lowest relapse risk was seen in patients with ATRA and chemotherapy maintenance.
ATRA plus ATO (0.3 mg/kg IV on days 1 and 5 of cycle one and 0.25 mg/kg twice weekly in weeks 2�8 or until clinical remission)
The combination of ATRA with chemotherapy improves long-term survival and results in 85-90% complete remission rates.
The most effective and least toxic induction chemotherapy combination with ATRA has not been established.
Currently, the three-drug regimen of ATRA 45 mg/m2 daily given 15 days every 3 months, oral PO) 6-MP 60 mg/m2 once daily, and methotrexate 20 mg/m2 PO once weekly are administered for 2 years, as maintenance therapy.
Monitoring is usually done by reverse transcription polymerase chain reaction (RT-PCR) assay for the PML-RARA fusion transcript.
All-trans retinoic acid (ATRA) binds to PML-RAR? and inhibits its antidifferentiation transcriptional activity.
ATRA induces degradation of the PML-RARA oncoprotein.
ATRA is a vitamin A derivative directed at a PML-RAR fusion transcript.
ATRA does not directly kill malignant cells, but induces terminal differentiation of malignant myeloid cells into mature neutrophils.
Early initiation of ATRA therapy is of paramount importance at the first suspicion of diagnosis to prevent catastrophic bleeding into the lungs or intracranially and early mortality of approximately 10%.
Early mortality and secondary malignancies affect the prognosis of APL adversely.
Early mortality results from complications: hemorrhage, differentiation syndrome, and infections.
Early mortality rates vary from 6% to 8% in clinical trials, 19% to 26% in single-institution studies, and up to 30% in population-based studies.
Retinoic acid is the treatment of choice as it overcomes differentiation blockade and restores normal granulopoiesis.
Treatment with retinoic acid alone results in the development of resistance.
Disease free survival rates of 67%-84% at 2 to 4 years when chemotherapy and all-trans retinoic acid (ATRA) are used.
ATRA 45mg/meter squared/ day orally until complete remission plus an anthracycline results in 84% complete remission rate.
ATRA in combination with cytotoxic chemotherapy limited to anthracyclines alone excluding cytarabine and chemotherapy based maintenance is present standard of care.
The cure rate with all-trans-retinoic acid plus an anthracycline-based chemotherapy is at least double, if not more than 4 other subtypes of AML who achieve complete remission.
This type of leukemia is the most durable of all acute leukemias in adults.
The induction mortality rate is approximately 10%-25% due to the APL coagulopathy.
Leukemic promyelocytes release substances that activity the coagulation cascade affecting the prothrombotic and fibrinolytic pathways.
Patients treated in the induction phase have a predisposition to clots but of greater likelihood of bleeding.
Management strategy includes aggressive correction of coagulopathy using blood products including platelet transfusions to maintain a platelet count greater than 50,000, fresh frozen plasma to correct coagulation factor deficiencies, to maintain a close to normal prothrombin time and partial thromboplastin time and cryoprecipitate to maintain fibrinogen greater than 150 mg/dL.
WBC counts of greater than 10,000mm3 associated with increased risk of early death, and increasing age less so.
Early deaths occur in 15-20% in individuals over the age of 60 years.
The most common causes of early death in APL are hemorrhage, differentiation syndrome (DS), and infection.
APL differentiation syndrome refers to a rapid rise in WBC count during APL treatment, resulting in hyperleukocytosis.
APL differentiation syndrome is linked to a lower serum albumin level.
High peripheral blood blasts, and coagulopathy increase the risk of death.
ATRA combined with an anthracycline based chemotherapy for induction, followed by two years of consolidation and then maintenance is the standard of care.
Arsenic trioxide has a high complete remission rate in relapsed acute promyelocytic leukemia.
Arsenic trioxide has a complete remission rate of 86% in 197 patients (Ghavamzade A et al).
Arsenic trioxide in the above study was associated with a 5 year disease free survival of 67% and the patients with more than one consolidation course of treatment-5 year disease-free survival approximated 90%.
Arsenic trioxide more effective than all-trans retinoic acid (ATRA) in reducing the degree of minimal residual acute promyelocytic leukemic cells.
Arsenic trioxide-as a single agent induces molecular remission in almost all patients treated at relapse with APL.
Arsenic trioxide in newly diagnosed patients using PCR testing the number of PML/RAR? transcript copies is significantly lower than with ATRA.
In a study of 73 patients with untreated acute promyelocytic leukemia treatment resulted in a complete remission of 90%.
Arsenic trioxide has an 86% complete remission rate in newly diagnoses patients, and when used for consolidation and maintenance is very effective in maintaining molecular remission.
Single agent arsenic trioxide associated with limited toxicity, with infrequent neutropenia.
ATRA reduces the risk of bleeding and coagulation problems.
ATRA should be administered at the first suspicion of the diagnosis, even before definitive diagnosis is made by cytogenetic or more like to the studies.
Patients require aggressive blood product management with cryoprecipitate and platelet support to maintain fibrinogen levels and platelet counts that adequate levels.
Management includes detection and treatment of infections with appropriate intravenous antibiotics, management of fluid status to avoid excessive fluids and maintaining adequate hydration, and prevention and treatment of the tumor lysis syndrome using fluids and allopurinol.
Age greater than 55-60 years is associated with a less favorable prognosis with a higher early death rate.
Elderly patients with good clinical conditions should receive the same treatment as younger patients, with a small dose reduction.
SEER results of a New York State Cancer Registry study revealed 10-20% of patients die within one month of diagnosis and more than 25% of patients died as a result of this process.
White blood cell count greater than 10,000 indicates a group of patients with whom the risk of relapse is elevated.
Higher risk of relapse and higher WBC counts include expression in APL blasts of antigen CD34, the neural adhesion molecule (CD56), and the T cell antigen CD2.
In patients with a very high white count administering high dose or intermediate dose cytosine arabinoside in consolidation, and the use of arsenic trioxide in consolidation therapy may decrease the relapse rate in this high risk group.
The highest risk of relapse is in the first 2 years.
There is a question of whether maintenance therapy should be done in patients who achieve a complete molecular remission at the end of consolidation.
Patients have resistant APL if they have not achieved complete molecular remission at the end of consolidation therapy.
Individuals have relapsed disease if they achieve molecular remission, but monitoring by RT-PCR assay shows positivity on consecutive samples.
Relapse disease can occur in up to 30% of patients.
Arsenic trioxide seems to be a better agent in APL than ATRA.
Once the diagnosis of APL is suspected in patients with white blood counts greater than 10,000 per microliter the immediate administration of ATRA and arsenic trioxide plus an anthracycline and cytosine arabinoside should be considered.
The combination of arsenic trioxide and all trans retinoic acid is associated with a two-year event free survival of 97% and a disease-free survival of 97% in standard risk patients, and is now standard of care for standard risk patients and it utilizes no chemotherapy(Lo-Coco F et al).
Chemotherapy agents utilized include, idarubicin.
Chemotherapy-free regimens are better tolerated without hair loss, less risk of infection, and other chemotherapy side effects
ATRA and arsenic administration typically used in low risk patients, and the addition for high risk patients of a third drug is considered.
ATRA and arsenic trioxide is a feasible treatment for low risk and high-risk patients with acute promyelocytic leukemia with a high cure rate and less relapse than, and survival not different from ATRA and idarubicin, with lowER incidence of liver toxicity.
Low-risk patients account for 2/3 of patients with APL.
In addition to the above the immediate administration of dexamethasone should be given to reduce the incidence of the APL differentiation syndrome, an adverse affect of both arsenic trioxide and ATRA (Tallman MS et al).
Rapid diagnosis and implementation of therapy is essential and hospitals should have adequate supplies of arsenic trioxide and ATRA on hand.
Prophylactic administration of platelets and clotting factors is essential.
Arsenic trioxide is an intravenous drug given daily for a prolonged period.
Arsenic trioxide is administered 5 days a week one month on and one month off, for a total of 8 months.
ATO is recommended in relapsed or refractory APL, showing shows high antileukemic activity, especially for patients who have a relapse within 1 year of receiving ATRA: complete remission rates of 52-96% with ATO monotherapy in relapsed APL.
ATO is well tolerated in elderly persons and has antileukemic effects at low doses.
Adverse effects of ATO include:
Prolongation of the QTc interval
Hepatotoxicity
Nausea and vomiting
Fluid retention
Itching and rash
Gemtuzumab ozogamicin is highly effective in molecular or overt relapsed disease.
Gemtuzumab or anthracyclines can be used to control the leukocyte count if necessary.
Patients with molecular or hematologic relapse after consolidation should be crossed over according to the previously used treatment.
Patients who previously received ATRA plus ATO should
receive ATRA plus chemotherapy and vice versa.
Autologous hematopoietic stem cell transplantation is performed in patients who have achieved second molecular remission, have negative MRD, or are have negative results on polymerase chain reaction (PCR) testing.
The patient’s individual genetic variants of APL should inform treatment: Patients with ATRA-sensitive variants should be treated with ATRA and anthracycline chemotherapy, while those with ATRA-resistant variants might not benefit from ATRA treatment.
Bone marrow transplantation (BMT) should be offered to those with relapsed APL.
Patients who achieve molecular remission with salvage therapy should be offered high-dose chemotherapy, followed by autologous stem cell transplantation (SCT) for consolidation.
Patients who have persistent molecular or hematologic disease after salvage therapy should be offered allogeneic SCT if they have a good performance status and an HLA-matched donor can be found.
Three-year overall survival is 73% for autologous stem cell transplant and 61% for allogeneic stem cell transplant.
No risk factor has been identified for prevention of a primary disease.
Secondary APL may follow exposure to chemotherapy agents, particularly mitoxanthone or to radiotherapy.
Secondary APL accounts for 10-20% of cases.
The disease and outcomes for secondary APL are similar to those of primary APL.
Treatment for secondary APL is similar to that of primary disease.