
 VEXAS syndrome is an adult-onset autoinflammatory disease primarily affecting males.
VEXAS syndrome is an adult-onset autoinflammatory disease primarily affecting males.
VEXAS syndrome is caused by a somatic mutation of the UBA1 gene in hematopoietic progenitor cells.
The UBA1 gene variant, reduced function of the UBA1 enzyme, which facilitates protein ubiquitination a post translational modification process, essential for cell signaling and protein degradation.
Patients experience autoinflammationwith recurrent episodes of inflammation in various organs.
The name VEXAS is an acronym derived from the features of disease:
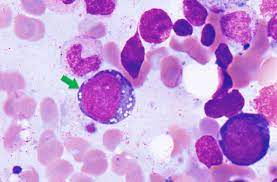
V: Vacuoles are often identified in the bone marrow stem cells of patients presenting with VEXAS.
E: The E1 ubiquitin conjugating enzyme encoded by the UBA1 gene is mutated.
X: The mutated UBA1 gene is located on the X-chromosome and thus the disease is almost exclusively found in individuals with XY chromosomes and thus said to be X-linked.
A: Autoinflammatory conditions
S: The mutations which cause VEXAS are somatic: they are acquired throughout life, not inherited, and are not passed on to offspring.
A somatic gene variant that is not inherited, and that the VEXAS syndrome predominantly affects people older than 50 years.
The prevalence is uncertain, but its a exome sequencing study on DNA extracted from peripheral blood reported that 1 in 4269 men older than 50 had a UBA1 variant associated with the VEXAS syndrome, although not all patients had symptoms.
VEXAS syndrome begins in late adulthood, typically after the age of 50.
It causes both autoinflammatory and hematologic symptoms.
Patients with the VEXAS syndrome have rheumatologic, hematologic, dermatologic, and pulmonary manifestations attributed to inflammation.
Skin lesions include vasculitis, neutrophilic dermatoses, erythema nodosum.
Patients may experience weight loss, recurrent fever, orchitis, hepatosplenomegaly, orbital inflammation with uveitis, scleritis, episcleritis, sensorineural, hearing loss and inflammatory arthritis.
sweet syndrome, giant cellarepteritid, polyarteritis nodosa, relapsing polychondritis, myelodysplastic, syndromes, and multiple myeloma have been reported.
The diagnosis should be considered in men with late onset undiagnosed, inflammatory syndromes with associated hematologic abnormalities.
Fever and rashes resembling those seen in Sweet syndrome are common signs.
It is associated with other autoinflammatory conditions: periorbital angioedema, uveitis and scleritis, relapsing polychondritis, polyarteritis nodosa, giant cell arteritis and lung involvement.
Hematologic manifestations include macrocytic anemia, a low platelet count, and a predisposition towards developing hematologic malignancies, especially myelodysplastic syndrome.
Bone marrow examination reveals abnormal vacuoles in precursor cells of the myeloid and erythroid lineages.
Lymphopenia occurs in approximately 80% and thrombocytopenia and 50% of patients.
On bone marrow examination more than 10% cytoplasmic values in myeloid and erythroid precursors and is an indication for UBA1 testing.
VEXAS syndrome becomes increasingly severe over time.
Diagnosis is made by identifying UBA1 gene variants with PCR, or tissue biopsy in a patient with VEXAS syndrome symptoms.
VEXAS syndrome has a high mortality rate.
Treatment: high-dose corticosteroid therapy.
Tapering of steroids may cause symptoms to recur.
Five-year survival of approximately 63%.