 Primary biliary cholangitis (PBC), previously known as primary biliary cirrhosis.
Primary biliary cholangitis (PBC), previously known as primary biliary cirrhosis.
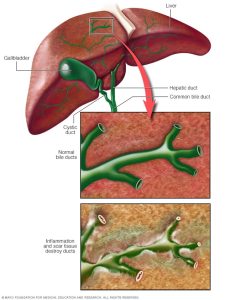
Primary biliary colitis is a chronic cholestatic autoimmune disease with destruction of small intrahepatic biliary epithelial cells.
Autoimmunity may play a role in the development of primary biliary cirrhosis with 53% of patients having at least one additional autoimmune illness.
It is caused by genetic, epigenetic and environmental factors.
Characterized by slow destruction of small intrahepatic bile ducts, portal inflammation and scarring with development of cirrhosis and hepatic insufficiency, and accumulation of toxic bile acids.
The process leads to the death of biliary epithelial cells in small bile ducts.
Overtime the number of bile ducts decrease ultimately resulting in cholestasis.
It causes bile and other toxins to build up in the liver, with cholestasis, information, and biliary fibrosis, which can progress to cirrhosis and liver failure, and death.
Secondary sclerosing cholangitis (SSC) is a chronic cholestatic condition that leads to progressive hepatic fibrosis and large-duct irregular biliary stricturing and dilation.
The cholangiographic appearance of SSC is similar to PSC.
A nonsuppurative inflammatory destructive process of medium sized intrahepatic bile ducts.
Pruritus, and fatigue are common debilitating symptoms.
Involves the small pulmonary arteries which have vascular proliferation and remodeling.
The process can be associated with pulmonary hypertension.
A chronic granulomatous cholangitis.
Most common autoimmune liver disease.
Cirrhosis develops after many years.
Chronic cholestatic liver process that affects primarily middle aged women.
Typically involves women in the fifth or sixth decade of life who presents with an elevated alkaline phosphatase.
Family and twin studies suggest a significant genetic predisposition.
Concordance among monozygotic twins is 60%, with sibling relative risk of approximately 10.5 (Jones DE).
Existence of other autoimmune disease in patients and the increased prevalence in families suggest a genetic influence.
Female:male ratio 6:1-9:1.
Approximately 3500 cases per year in the US and 47,000 probable cases among the white population(Kim WR).
Prevalence in the U.S. estimated at 150-400 per million women.
Affects as many as 1 in 1000 women over the age of 40.
Frequency: one and 3 to 4000 people.
Age of onset between 20 and 80 years, with peak ages 40-50.
8% of patients have associated scleroderma.
PBC can also be associated with metabolic bone disease, steatorrhea, and deficiencies of fat-soluble vitamins.
Autoimmune thyroid disease such as Hashimoto thyroiditis, goiter and rarely Graves’ disease associated in approximately 15 to 20% of cases (Culp, KS).
Concomitant autoimmune diseases are common in patients with PBC: Sjogren syndrome, thyroid disease, limited cutaneous scleroderma, and rheumatoid arthritis.
Rheumatoid arthritis, Raynard phenomenon, esophageal dysmobility, sclerodactyly, telangiectasia syndrome, pernicious anemia, celiac disease, inflammatory bowel disease and pulmonary fibrosis have been associated with PBC.
Autoimmune disorder triggered by environmental toxins, chemical exposure or infectious agents that cause a self-perpetuating process.
Associated with certain HLA haplotypes suggesting genetic susceptibility.
Antimitochondrial antibodies characteristic and present in 95% of patients.
Antimitochondrial antibodies are specific for the E2 subunit of the private dehydrogenase complex.
There is an accumulation of auto reactive T lymphocytes in the liver.
Suggested that CD4+ T helper lymphocyte helper cells are responsible for the pathogenesis of the autoimmune cholangitis.
Lipid abnormalities are common in PBC.
Mild elevations in low-density lipoprotein (LDL) with more marked elevations in high-density lipoprotein (HDL) are common in early PBC, whereas more prominent elevations in LDL and lower HDL levels are noted in later stages of the disease.
Lipoprotein-X an atypical lipoprotein found in primary biliary cirrhosis and can account for a substantial fraction serum cholesterol.
Lipoprotein-X it fears would make the panel measurements and falsely increases calculated LDL and falsely alters HDL levels.
In patients with hyperlipidemia treatment of the cholestasis is indicated with the use of ursodeoxycholic acid (UDSA).
Despite the presence of hyperlipidemia no evidence exists for an increased prevalence of atherosclerotic disease or cardiovascular related mortality in patients with primary biliary cirrhosis.
The hyperlipidemia in PBC does not appear to confer excess cardiovascular risk, due, in part, to the fact that elevated levels of lipoprotein X (LP-X) that comprise a substantial proportion of cholesterol in patients with advanced PBC confers atheroprotection by inhibiting oxidation of LDL.
Antinuclear antibodies found in approximately 50% of patients.
Anti mitochondrial antibody (AMA) is a disease-specific antibody for PBC, present in 95% of patients with the condition.
Associated with Hashimoto’s thyroiditis.
Sjogren syndrome seen in up to 70%-80% of cases if specific testing is done.
Gallstones seen in about 30% of cases.
Patients present with fatigue, pruritus, jaundice, hepatomegaly, splenomegaly and hyperpigmentation.
Jaundice is a late finding, and early on the only changes may be abnormal liver function tests.
Approximately 60% of patients with PBC are asymptomatic at the time of diagnosis, which is made on the basis of abnormal liver biochemical tests.
Up to 80% of patients present with fatigue, which may be debilitating.
Pruritus may interfere the quality of life and treatment options include cholestyramine, antihistamines, opioid antagonists, rifampin, and sertraline.
Pruritus occurs in 20 to 70% of cases, and can develop at any stage of the disease.
Pruritus does not correlate with progression of liver disease.
Pruritus is mild to moderate intensity, and may improve or disappear as the disease advances.
Pruritus correlates with with fatigue and impacts quality of sleep.
Pruritus is rarely severe, and is characteristically intermittent, worse at night, and improves during summer.
With refractory pruritus liver transplant may be effective.
The development of hepatomegaly is typical.
Laboratory abnormalities include elevation in ALP with normal or mild elevations in serum aminotransferases.
Elevation of serum bilirubin suggests advanced disease and constitutes a poor prognostic sign.
May be associated with osteoporosis and hypercholesterolemia.
Bilirubin inhibits osteoblast function and elevated bilirubin may account for impaired bone formation and the presence of osteoporosis.
Fat soluble vitamins, calcium and vitamin D supplementation are recommended.
14% of first-degree relatives have an autoimmune disorder.
Median survival for patients with asymptomatic disease versus 8 years for patients with symptoms.
Most patients are asymptomatic at the time of diagnosis, and this occurs because of the use of chemistry profiles as part of a general medical evaluation.
Approximately 60% of patients are asymptomatic at the time of diagnosis.
Common presenting symptoms in symptomatic patients with PBC are fatigue and pruritus.
Findings on physical examination in PBC include: hepatomegaly, skin hyperpigmentation, jaundice, xanthomas, or stigmata of cirrhosis and portal hypertension in more advanced stages.
In situations where the clinical pattern is consistent with cholestatic liver disease confirmatory liver biopsy is not necessary for final diagnosis.
An antimitochondrial antibody test may be the single diagnostic test needed for the diagnosis in the appropriate clinical pattern.
Liver biopsy may be useful in patients who have a negative antimitochondrial antibody .
Patients with a negative antimitochondrial antibody test have similar clinical course, prognosis and response to treatment as those with a positive test.
A progressive loss of intrahepatic bile ducts with progression to advanced fibrosis, cirrhosis and liver failure.
Cause of death in such patients is liver failure.
Median survival used to be 10 years and now approaches normal life span when treated early.
Clear benefits from prednisone and methotrexate have not been shown.
Only recommended treatment is the use of ursodeoxycholic acid (UDCA) 13-15 mg/kg per day.
Cholestyramine may relieve pruritus.
UDCA should be utilized in all newly diagnosed patients.
Men tend to be older, and have higher bilirubin levels at the time of diagnosis than women.
Men are less likely than women to have a favorable response to ursodeoxycholic acid, the first line therapy.
Treatment with ursodeoxycholic acid results in normalization of alkaline phosphatase level, gammaglutamyl transferase level, and the alanine aminotransferase level in approximately 60% of patients by two years.
Treatment with ursodeoxycholic acid is associated with a significant increase in five and 10 year survival.
Treatment success is determined by the levels of serum alkaline phosphatase, and billirubin present after one year of therapy.
Up to 40% of patients have an adequate response toursodeoxycholic, and are at high risk for progression of disease.
Once liver failure develops, hepatic transplant is the only available option.
Liver transplantation for primary biliary cholagitis is considered for severe liver disease, as well as for portopulmonary hypertension.
In patients with non-cirrhotic stage disease , treatment with UDCA is associated with a survival that does not differ from an age and sex-matched general population (Poupon RE).
UDCA delays disease progression and prolong survival, particularly in patients whose alkaline phosphatase is lower than twice the upper limit of normal after six months of treatment, and in patients with early-stage disease 90 (Anguuulo P, Corpechot C).
Ursodeoxycholic acid (UDCA) is the only FDA approved drug for treatment.
In a review of 16 randomised clinical trials with a total of 1447 patients with the primary outcome measures of mortality or liver transplantation: UDCA showed a reduction in liver biochemistry, jaundice, and ascites, but did not demonstrate any benefit on mortality and mortality or liver transplantation.
Up to 40% of patients treated with Ursodeoxycholic acid have persistently elevated, alkaline phosphatase level, bilirubinm level, or both, portending disease progression.
In advanced disease UDCA associated with a poorer prognosis.
Immunosuppressive agents have not been shown to be beneficial.
Seladelpar, a peroxisome proliferator – activated, receptor delta agonist has biochemical response and alkaline phosphatase normalization that is significantly greater than with placebo.
Seladelpar significantly decreases pruritus.
Obeticholic is a second line therapy for primary biliary cholangitis(abile acid analogue).
Treatment with elafibranor resulted in significantly greater improvements in relevant biochemical indicators of cholestasis than placebo.
Elafibranor, a dual peroxisome proliferator – activated receptor delta agonist the decreases the toxic effects of bile acid and inflammation.