 Neuroendocrine tumors are cancers that begin in specialized cells called neuroendocrine cells.
Neuroendocrine tumors are cancers that begin in specialized cells called neuroendocrine cells.
Neuroendocrine tumors (NETs) are neoplasms that arise from cells of the endocrine and nervous systems.
Cells that can give rise to NETs are present in endocrine glands, most commonly Kulchitsky cells or similar enterochromaffin-like cells, that are relatively more common in the gastrointestinal and pulmonary systems.
Neuroendocrine cells have traits similar to those of nerve cells and hormone-producing cells.
Neuroendocrine tumors are rare and can occur anywhere in the body.
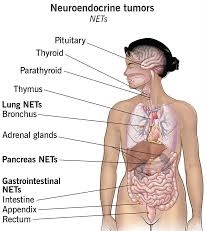
NETs include tumors of the gastrointestinal tract and of the pancreatic islet cells, thymus and lung tumors, and medullary carcinoma of the parafollicular cells of the thyroid.
Tumors with similar cellular characteristics are found in the pituitary, parathyroid, and adrenomedullary glands.
Pituitary gland: Neuroendocrine tumor of the anterior pituitary
Thyroid gland: Neuroendocrine thyroid tumors, particularly medullary carcinoma
Parathyroid tumors
Thymus and mediastinal carcinoid tumors
Pulmonary neuroendocrine tumors
bronchus
pulmonary carcinoid tumors: typical
carcinoid (TC; low-grade); atypical
carcinoid (AC; intermediate-grade)
small-cell lung cancer (SCLC)
large cell neuroendocrine carcinoma of the lung (LCNEC)
Extrapulmonary small cell carcinomas (ESCC or EPSCC)
Gastroenteropancreatic neuroendocrine tumors (GEP-NET)
Foregut GEP-NET encompasses not only NETs of the stomach and proximal duodenum, but also the pancreas, and even thymus, lung and bronchus.
Pancreatic endocrine tumors
Midgut GEP-NET-from distal half of 2nd part of the duodenum to the proximal two-thirds of the transverse colon) appendix, including well differentiated NETs (benign); well differentiated NETs (uncertain malignant potential); well differentiated neuroendocrine carcinoma (with low malignant potential); mixed exocrine-neuroendocrine carcinoma (goblet cell carcinoma, also called adenocarcinoid and mucous adenocarcinoid)
Hindgut GEP-NET Liver and gallbladder
Adrenal tumors, particularly adrenomedullary tumors
Pheochromocytoma
Peripheral nervous system tumors, such as: Schwannoma paraganglioma neuroblastoma
Breast
Genitourinary tract urinary tract carcinoid tumor and neuroendocrine carcinoma ovary
neuroendocrine tumor of the cervix
Prostate tumor with neuroendocrine differentiation
Merkel cell carcinoma of skin
Several inherited conditions:
multiple endocrine neoplasia type 1 (MEN1)
multiple endocrine neoplasia type 2 (MEN2)
von Hippel-Lindau (VHL) disease
neurofibromatosis type 1
tuberous sclerosis
Carney complex
The term neuroendocrine tumor generally refers to a well differentiated disease, while neuroendocrine carcinoma refers to poorly differentiated disease: Neuroendocrine neoplasms is an umbrella term for both.
NETs are thought to arise from cells throughout the diffuse endocrine system.
Most neuroendocrine tumors occur in the lungs, appendix, small intestine, rectum and pancreas.
NETs most commonly occur in the intestine, where they are often called carcinoid tumors, but they are also found in the pancreas, lung and the rest of the body.
Neuroendocrine lesions are graded histologically by their markers of cellular proliferation, rather than cellular differentiation.
Grading: By mitosis and Ki-67%.
GX Grade cannot be assessed
G1 < 2 < 3%
G2 2 to 20 3–20%
G3 > 20 > 20%
If mitotic count and Ki-67 are discordant, the figure which gives the highest grade is used.
G1 and G2 neuroendocrine neoplasms are referred to as neuroendocrine tumors (NETs) – formerly called carcinoid tumors.
((Carcinoid tumors))most commonly affect the small bowel, particularly the ileum, and are the most common malignancy of the appendix.
Many carcinoids are asymptomatic and are discovered only upon surgery for unrelated causes.
Coincidental carcinoids are common; one person in ten has them.
Many carcinoid tumors do not cause symptoms even when they have metastasized.
Other tumors even if very small can produce adverse effects by secreting hormones.
Ten per cent or less of carcinoids, primarily some midgut carcinoids, secrete excessive levels of a range of hormones, most notably serotonin (5-HT) or substance P, causing a constellation of symptoms called ((carcinoid syndrome)).
Carcinoid syndrome symptoms:
flushing
diarrhea
asthma or wheezing
congestive heart failure (CHF)
abdominal cramping
peripheral edema
heart palpitations
A carcinoid crisis presents with profound flushing, bronchospasm, tachycardia, and widely and rapidly fluctuating blood pressure with large amounts of hormone are acutely secreted.
Chronic exposure to high levels of serotonin causes thickening of the heart valves, particularly of the tricuspid and the pulmonic valves
Chronic exposure to high levels of serotonin over a long period can lead to congestive heart failure.
Valve replacement is rarely needed in carcinoid syndrome.
The excessive outflow of serotonin can cause a depletion of tryptophan leading to niacin deficiency, and thus pellagra.
((Pellagra)) is associated with dermatitis, dementia, and diarrhea.
Other hormones can be secreted by neuroendocrine tumors: most commonly growth hormone that can cause acromegaly, or cortisol, that can cause Cushing’s
Hormone release in carcinoid tumors is occasionally triggered by diet, alcohol, surgery, chemotherapy, embolization therapy or radiofrequency ablation.
G3 neoplasms are called neuroendocrine carcinomas (NECs).
NETs share common features: appear similarly, have special secretory granules, and often produce biogenic amines and polypeptide hormones.
Estimated incidence is 6.98 cases per hundred thousand people and is increasing with the prevalence estimated at greater than 170,000 in the US.
Misdiagnosis was common (44%) in these patients, and mean time to diagnosis was around 5 years.
Because NETs can present in different areas of the body, lead to diverse symptoms, and are rare, timely diagnosis and management are challenging.
Rates are increasing rapidly, potentially due to improved awareness of the condition and diagnostic tools.
There are many types of neuroendocrine tumors.
Some grow slowly and some grow very quickly.
Some neuroendocrine tumors produce excess hormones and are functional neuroendocrine tumors.
The World Health Organization (WHO) classification scheme places neuroendocrine tumors into
categories, which emphasize the tumor grade rather than the anatomical origin:
well-differentiated neuroendocrine tumors, with benign and with uncertain behavior
well-differentiated (low grade) neuroendocrine carcinomas with low-grade malignant behavior
poorly differentiated (high grade) neuroendocrine carcinomas, which are the large cell neuroendocrine and small cell carcinomas.
Grading such tumors into one of these categories depends on well-defined histological features: size, lymphovascular invasion, mitotic count, Ki-67 labelling index, invasion of adjacent organs, presence of metastases and whether they produce hormones.
Traditionally, neuroendocrine tumors are classified by their anatomic site of origin.
Most often they are located in the intestine, pancreas or the lungs.
Up to 25% of NETs are functional and releasendistinctive hormones into the bloodstream, including amines, polypeptides, and prostaglandins.
Others tumors don’t release hormones or do not release enough to cause symptoms and are nonfunctional neuroendocrine tumors.
Neuroendocrine tumors are highly vascular tumors and vascular endothelial growth factor is a key driver of angiogenesis.
Neuroendocrine tumors are classified, according to the site of origin, morphologic features, grade, and differentiation.
NET’s are subclassified as well differentiated or poorly differentiated.
The grade is based on the KI-67 proliferative index and/or mitotic rate.
The grade may be low with the KI-67 in less than 3%, moderate with a KI-67 of 3 to 20%, or high with KI-67 greater than 20%, related to grades 1,2, and 3 respectively.
The term neuroendocrine tumor refers to a well differentiated neoplasm, whereas neuroendocrine carcinoma, indicates a poorly differentiated carcinoma.
Neuroendocrine tumor grade 3 covers all highgrade neuroendocrine malignanciess with a KI 67 higher than 20%: Grade 3 NET is well differentiated and neuroendocrine carcinoma if it is poorly differentiated.
G3 NET makes up approximately 18% of all Neuroendocrine neoplasms.
The most common primary organ of an NET G3 is the pancreas (65%).
Pancreatic NETs are associated with a relatively high expression of VEGF receptor 2, 3, platelet derived growth factor receptor.
Diagnosis and treatment of neuroendocrine tumors depend on the type of tumor, its location, whether it produces excess hormones, how aggressive it is and whether it has spread to other parts of the body.
Types of NETs
Adrenal cancer
Carcinoid tumors
Merkel cell carcinoma
Pancreatic neuroendocrine tumors
Paraganglioma
Pheochromocytoma
NET‘s may arise in the context of inherited genetic syndromes, including multiple endocrine neoplasia types one, two, and four and with succinate dehydrogenase mutations.
NET‘s have been associated with Von Hippel-Lindau disease, tuberous sclerosis complex, neurofibromatosis, MEN1, and MEN2,
Symptoms
Neuroendocrine tumors don’t always cause signs and symptoms at first.
Symptoms experienced depend on the location of the tumor and whether it produces excess hormones.
Patients have functional tumors when associated with hormonal symptoms, and for those without symptoms they have non-functional tumors.
Symptoms include intermittent flushing and diarrhea in patients with gastrointestinal NET‘s, bronchospasm and wheezing in lung NETs, hypertension in patients with pheochromocytoma or paragangliomas, and symptoms attributable to secretion of insulin, glucagon, gastrin, and other peptides in patients with pancreatic NETs.
In general, neuroendocrine tumor signs and symptoms might include:
Pain from an expanding tumor.
A growing mass you can palpate.
Fatigue
Losing weight without trying
Neuroendocrine tumors that produce excess hormones
Functional neuroendcrine tumors might cause:
Skin flushing
Diarrhea
Frequent urination
Increased thirst
Dizziness
Shakiness
Skin rash
Diarrhea with NET‘s can be multifactorial: exocrine pancreatic insufficiency, short bowel syndrome, bacterial overgrowth, and bile acid malabsorption.
Exocrine pancreatic insufficiency develops in the majority of patients on somatostatin analogues.
NETs staining with synaptophysin and chromogranin and have epithelial markers of cytokeratin and CAM.
hormonally inactive peptides, such as chromogranin A and pancreatic polypeptide conserve his tumor markers for non-functioning NETs.
Evaluation of serotonin secretion using 24 hour urine or plasma 5-HIAA is indicated for carcinoid syndrome.
syndrome.
Bowel obstruction can occur, due to fibrosing effects of NET secretory products with an intense desmoplastic reaction at the tumor site, or the mesentery.
((Pancreatic neuroendocrine tumors)) are often referred to as islet cell tumors.
About 95 percent of pancreatic tumors are adenocarcinoma; only 1 or 2% of clinically significant pancreas neoplasms are GEP-NETs.
NETs are grouped in three grades based on the KI-67 proliferative index and mitotic rate, which have prognostic and treatment applications.
NETs are characterized as well differentiated tumors, and poorly differentiated tumors are classified as Neuroendocrine carcinomas.
Tumorigenesis and molecular pathogenesis a highly variable depend on the tumor location.
Mutations of chromatin remodeling MEN 1, DAXX, and ATRX are common in pancreatic NETs and correlate with poor prognosis.
Small bowel NETs have mTOR pathway alterations in approximately 1/3 of tumors and gain of function in chromosomes 4, 5, and 14 with loss of chromosome 18 and in activation of CDKN 1/APC.
In small bowel NETs pathway alterations occur in approximately 1/3 of tumors.
Small bowel NET‘s are mostly sporadic.
NET‘s are classified as functional or non-functional.
25% of all NETs are functional.
Treatment is focused on controlling both tumor growth and symptoms of functional tumors.
Curative resection is generally recommended whenever possible.
Bronchial/pulmonary NETs: surgical resection with curative intent is the preferred approach for stages I through III when possible.
All poorly differentiated neuroendocrine carcinomas are grade 3.
Imaging is most effective with somatostatin receptor scintography with PET/CT scans using gallium68 and Cu 64.
GA-DOTATE positron emission tomography/CT has become the preferred imaging owing to its superior ability in detection of gastrointestinal pancreatic- NETs when compared with standard anatomic and octeotide imaging.
These have a different prognosis and different clinical approach to treatment.
Treatment decisions depend upon the disease biology, symptom burden, disease extent, patient comorbidities, and patient preference.
Surgery is considered with curative intent at an early stage or in localized NETs.
If possible, localized low and intermediate grade ET‘s should be respected with negative margins and lymph node dissection should be considered in most cases.
Median time for recurrence is 8.7 years in small intestine NETs and 7.2 years for pancreatic NETs: suggesting surveillance particularly with MRI for younger patients to reduce radiation exposure.
Lver directed therapies including surgical resection, hepatic arterial embolization, percutaneous thermal ablation may be considered for patients who have progressed on systemic therapy and have symptomatic disease.
Observation for early nonfunctioning pancreatic, neuroendocrine tumors of 2 cm or less is supported by current guidelines.
Neuroendocrine tumors are one malignancy where surgery for advanced or metastatic disease is a consideration, when greater than 70-90% of the disease can be resected, or when tumor debunking of functional tumors is needed in symptomatic patients.
Not all patients with advanced disease require therapy and the disease can be observed with tumor markers and imaging studies to establish the burden of disease and its progression.
Functional NET tumors cause distinct syndromes such as carcinoid syndrome because of secreted peptides and neuroamines.
Patients with syndrome symptoms of hormone secretion often benefit from treatment with somatostatin analogues including octreotide, lanreotide, pasireotide and telotristat.
Peptide receptor radionuclide therapy exploits the expression of somatostatin receptors on the surface of NET cells and delivers a radionuclide at the cellular level.
Radio active labeled agents are composed of radionucleotide isotopes such as yttrium 90, or lutetium 77.
Overall regardless of the site of origin curative resection is recommended.
Adjuvant therapy has no role in the management of well differentiated NETs.
For bronchial/pulmonary NETs surgical resection with curative intent is preferred for stage is I-III, when possible.
Local regional bronchial and thymic NETs have a five-year survival rate after curative resection is high as 97%.
The survival of patients with atypical carcinoids and a higher T stage is inferior to patients with typical carcinoids and smaller primary tumors.
Targeted therapy with everolimus, radionuclide therapy, interferon, cytotoxic chemotherapy, and liver directed therapy for hepatic predominant disease.
Lu-177-Dotatate A peptide receptor radioclide therapy is available.
In patients with SSTR positive GEP-NETs LU-Dotatate has been shown to offer disease control and survival benefit.
Lu-177-Dotatate Dosing is every eight weeks for four treatments.
Gastroduodenal NETs categorized into three types.
Type one gastric NETs are associated with gastric achlorhydria and are the most benign type, constituting 70 to 80% of all gastric NETs.
Type 2=gastric NETs occur as part of multiple endocrine neoplasia 1 syndrome it is associated with pancreatic duodenal gasttinomas.
Type 3 gastric NETs are sporadic and occur in the absence of gastric achlorhydria or Zollinger-Ellison syndrome and may constitute up to 20% of all gastric NETs.
Most patients with gastric NETs have local or hepatic metastases at the time of presentation.
Surgical resection depends on the type and functional status of the tumor.
Active surveillance is reasonable in subcentometer gastric NETs.
Surgical resection of pancreatic NETs is the mainstay of therapy except for high-grade tumors, symptomatic or functional tumors, and tumors measuring at least 2 cm.
For patients with larger tumors in whom lymph node metastasis is strongly suspected a Whipple procedure is the preferred approach.
Surgical resection of primary disease and disease debulking along with systemic therapy may play a role in the management of symptomatic and functional pancreatic NETs.
Surgical resection is the cornerstone of treatment for midgut NETs involving the small and large bowel.
With appendiceal NETs appendectomy is preferred when the tumor is smaller than 1 cm and the right hemicolectomy is considered when tumor is a larger than 2 cm.
The liver is the most common site distant NET metastases, and hepatic metastases are a major predictor of survival in this disease.
Liver directed therapy is an option for patients with predominantly liver metastasis.
Systemic agents such as tyrosine kinase inhibitors and chemotherapy for pancreatic NETs and m-TOR inhibitors are considered.
Sunitinib, a multi targeted tyrosine kinase inhibitor can be used for advanced pancreatic neuroendocrine tumors with the response rate of 9%.
Everolimus is approved for neuroendocrine tumors with studies, showing a median progressive free survival of 11.0 versus 4.6 months in placebo studies.
80 to 90% of small bowel NETs and 60 to 70% of pancreatic NETs eventually metastasize to the liver.
Surgical resection of a primary tumor along with neuroendocrine liver metastases, when feasible, has been associated with prolonged progression free survival.
Segmental resection, radiofrequency ablation, and transarterial procedures with chemoembolization and transarterisl radio embolization are other alternatives that can provide durable disease control of liver disease.
In patients with liver metastasis liver transplant may be an option with a five year overall survival rate of 63%.
Somatostatin analogues, are typically the first line therapy to control the growth of a well differentiated NET.
Octreotide is approved for hormonal control of NETs, while lanreotide is approved for tumor control as well.
Luteum177 is a therapeutic consideration for somatostatin receptor positive gastroenteropancreatic NETs.
In the NETTER –1 trial Lutetium LU 177 in patients with advanced, somatostatin receptor positive midgut NETs that had progressed on standard octreotide therapy had a overall response rate of 18% with a 79% reduction in risk for death.
Chemotherapy with efficacy include: streptozotocin, temozolomide, 5FU, capecitabine and oxaliplatin.
Neuroendocrine tumor associated hormonal syndromes include insulinoma, gastrinoma (Zollinger Ellison syndrome, VIPoma, somatostatinoma and glucagonoma.
Management of NETs depend on factors related to the symptoms, pace of disease progression, and disease location.
Surveillance may be used in patients with low grade tumors and low tumor burden, who are asymptomatic, who have non-functional, stable, low-grade midgut tumors with a KI 67 index no higher than 10%.