 Accounts for 6%-10% of all new cases of NHL.
Accounts for 6%-10% of all new cases of NHL.
Its frequency is similar to that of non-cutaneous peripheral T cell lymphomas.
3-4000 cases per year in the U.S.
Median age of presentation is the mid 60s.
Annual incidence 0.4 per 100,000 persons.
More common in men than women (3:1) with a median age ranging from 60 to 70 years.
Incidence increasing.
A B-cell lymphoma, non-Hodgkin type.
Classified as an aggressive NHL.
Has the worst prognosis among B-cell subtypes.
A few patients have an indolent leukemia presentation with normal LDHN Lo KI-67.
Considered incurable.
Important factors for the development of other lymphomas such as familiar risk, immunosuppression, other immune disorders, chemical and occupation exposures and infectious agents have not been condensingly identified as those predisposing for mantle cell lymphoma, with the possible exception of family history.
Median age of patients is between 60 and seven years, similar to the median age of patients with diffuse large B cell lymphoma.
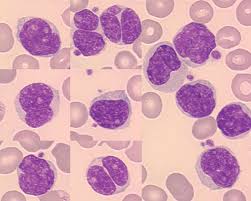
The malignant cells are small to medium size lymphocytes, there is scant cytoplasm, clumped chromatin, inconspicuous nuclei, and prominent nuclear clefts.
Mantle cell lymphoma growth patterns include diffuse and nodular.
Lambda light chains and more commonly rearranged than kappa light chains, and the tumor cells express IgM and IgD on the surface as well as CD5, characteristics they share with CLL.
The cells are negative for CD10and BCL-6 and usually do not expressed CD23.
In nearly all cases the tumor cells overexpress cyclin D1 that drive cells from the G1 phase into the S phase, SOX11 a transcription factor, and Bcl-2 and antiapoptosis protein.
Typical cells have a characteristic t(11; 14) translocation.
The malignment cell is often a pre-terminal center B cell and clonally rearranged immunoglobulin genes that are largely unmutated.
Characterized by the transposition of the BCL1 gene (11 q13) to a site downstream of the immunoglobulin heavy chain gene promoter leading to the pathognomonic the(11;14)(q13;q32) translocation, which leads to the constitutive overexpression of cyclin D1.
The t(11;14)(q13;q32) translocation is the hallmark of the disease juxtaposing the cyclin-D1 gene CCND1 at 11q13 with the immunoglobulin heavy chain gene (IgH) on the derivative chromosome 14, resulting in over expression of cyclin D1.
In rare cases of mantle cell lymphoma cycle in D1 is not expressed.
Often these tumor’s express cycling D2, D3, or E instead.
Characterized by CD5 positive CD23 negative follicular mantle B-cells.
Mantle cell lymphoma has a distinctive immunophenotype, typically positive for pan B-cell markers, CD5 and cyclin D1, but negative for CD10, CD23, and CD200.
CD23 expression linked to improved survival in MCL.
In a large cohort of patients with mantle cell lymphoma (MCL), CD23 expression was associated with significantly improved survival outcomes.
Mantle cell lymphoma has a distinctive immunophenotype, typically positive for pan B-cell markers, CD5 and cyclin D1, but negative for CD10, CD23, and CD200.
Some MCL cases have atypical immunophenotypic features, such as expression of CD10, CD23, or rarely CD200 or lack of expression of CD5.
Saksena and colleagues found that patients with CD23-positive MCL more frequently had bone marrow involvement 89% vs. 78%, a leukemic nonnodal presentation 42% vs. 11%, an elevated leukocyte count 33% vs. 18%, and stage 4 disease 87% vs. 77%
The researchers reported that CD23 expression was associated with significantly improved PFS and OS.
About 25% of cases may be t(11;14)negative and express cyclin D2 or D3, the expression of which may be driven by IG medited translocations (Wiodarska I).
Lesions that are t(11;14) negative have a predisposition for CNS involvement (Wiodarska I).
Characterized by cell cycle dysregulation and a defective DNA damage response pathway.
Markers of very high risk mantle cell lymphoma include blastoid and pleomorphic cytologic patterns, KI-67 index greater than 30%, and TP53 mutation or deletion.
Has the poorest long-term prognosis all the lymphoma subtypes.
TP53 gene aberrations are associated with poor prognosis.
Most patients with MCL present with palpable lymphadenopathy, with or without systemic symptoms.
Localized nodal or extranodal disease is a present in approximately 10% of patients and more than 80% of stage III or IV disease at diagnosis, frequently with bone marrow involvement.
Often involves extranodal sites, such as bone marrow, blood, nasopharyngeal and G.I. tract.
Generally aggressive and incurable.
About 30% of patients have fevers, sweats, or weight loss, but more patient report fatigue.
Bulky adenopathy is seen in approximately 25% of patients, with masses 10 cm or greater, and fewer than half of patients have an elevated LDH.
CNS involvement is rare at initial presentation, but is associated with a very short survival.
Five-year survivall rates range from 70% for those with limited stage disease to 50% for those having advanced disease.
Medium progression free survival of 16.6 months and overall survival of 4-5 years from first diagnosis.
Displayed initial response to therapy, disease progression is common and the rate of one year overall survival is only 22%.
Does have a spectrum of disease, with 10-15% of patients presenting with an indolent process with splenomegaly, no lymphadenopathy,, high white blood count, and cytogenetics which are not complex, making it much more stable genetically
Incurable by conventional chemotherapy.
Male to female ratio of 2-3:1: approximately 70% of cases occur in men.
Most patients are older and are unsuitable candidates for aggressive treatment or cannot tolerate stem cell transplantation.
No etiologic factors identified.
Occurs more frequently in the early mid 60s age group.
Median survival of approximately 4-5 years, if not eligible for transplant.
Many patients present with fever, sweats, weight loss and symptoms due to extra nodal disease.
Approximate 75% of patients are initially diagnosed with advanced stage III or IV disease.
Most common sites of disease include lymph nodes, spleen, Waldeyer’s ring and bone marrow.
Histologically nodes show diffuse (most common), nodular and mantle zone growth patterns.
Diagnosis usually made by lymph node biopsy of tissue from blood or bone marrow.
Mantle cell lymphoma cells are typically small-medium sized lymphoid cells with irregular nuclear contours.
Cells are reported to be small cell, pleomorphic/diffuse and blastoid types.
The pleomorphic and blastoid types are frequently associated with p53 mutation or deletion and have a poor outcome.
Cells are small to medium in size with clefted or folded nuclei, fine chromatin and small nucleoli.
Variants from classic changes include small cell, allomorphic and blastoid types.
Blastoid variants often associated with del17P.
Divided into extranodal, nodal and splenic types.
Monoclonal B cell population with expression of CD5, CD10, CD20, surface immunoglobulin M, and FMC7.
Cells overexpress cyclin D1.
Cyclin D1 negative case have been reported and have a similar morphology and gene expression and have overexpression of cyclin D2 and D3.
SOX11 is overexpressed.
Commonly associated with genetic instability with alterations of p53, P16, p21 or ATM.
Ki67 level is a surrogate marker.
Diagnosis confirmed by the t(11;14)(q13;q32)translocation.
Thought to derive from CD5+ antigen-naïve, pregerminal center B cells within the mantle zone surrounding normal lymphoid follicles.
CD10 and CD23 and BCL-6 typically absent.
PI 3-kinase/Akt prosurvival signaling pathway and is activated in the more aggressive blastoid form of disease.
Known as centrocytic lymphoma under the Kiel classification.
Ki-67 Index with a 30% or greater expression of mantle cell lymphoma cells by immuno chemistry is associated with shorter survival.
Median age at diagnosis 63 years.
In the NHL classification project has the worst 5 year survival of any lymphoma subtype.
The Mantle Cell Lymphoma International Prognostic Index (MIPI) recognizes poor prognostic factors such as age of 60 years of greater, impaired performance status, elevated LDH, and elevated total leukocyte count.
Recent studies indicate a median survival is 4.8 years, and the five year survival of 44%.
Associated with a high recurrence rate.
Patients with relapsed disease respond poorly to therapy and progress rapidly resulting in a median overall survival of one-two years.
Except for a small patient population eligible for an allogeneic stem cell transplant there is presently no recognized standard of care in relapsed patients.
Usually sensitive to initial treatment and subsequent relapses with rapid emergence of resistance resulting in a relatively short disease free survival time.
Considered noncurable.
Currently considered an aggressive intermediate grade lymphoma.
Occurs predominantly in men and commonly affects lymph nodes, spleen, bone marrow, peripheral blood, and the gastrointestinal tract.
Majority have advanced Ann Arbor stage III/IV disease with disseminated nodal and bone marrow involvement.
Approximately 90% present with advanced disease.
Gastrointestinal tract involved in more than 90% of patients and Waldeyer’s ring also commonly involved.
Gastrointestinal involvement most commonly includes the colon, ileocecal valve, and stomach.
Occasionally associated with intestinal lymphomatous polyposis.
Extranodal involvement can occur in the skin, breast, CNS lacrimal gland, genitourinary tract and eyelid.
May present with a leukemia phase and this subtype is associated with splenomegaly and CNS involvement with a worse prognosis and median survival of 12-18 months or less.
There is an association between Borrelia burgdoferi infections and this lymphoma.
Treatment:
A subgroup of patients do not require immediate therapy and can be safely observed.
Patients with splenomegaly, bone marrow involvement, and circulating lymphoma cells but don’t have lymphadenopathy are usually asymptomatic and do not require immediate therapy.
Patients with a nodal presentation but with low-volume disease and no symptoms can be safely observed.
Complete response rates with standard CHOP (cyclophosphamide/doxorubicin/ vincristine/prednisone) chemotherapy 10-60%, with further improvement with the addition of rituximab.
VR-CAP( bortezomib, Rituximab, cyclophosphamide, doxorubicin, and prednisone is associated with a significantly longer survival than R-CHOP.
Rituximab has limited activity as monotherapy and is more effective when used in combination with chemotherapy.
Median survival with CHOP 3 years and median time to treatment failure 15-18 months.
Initial therapy includes chemoimmunotherapy with intense therapy such as high-dose chemotherapy and hematopoietic transplantation for individuals who are candidates for transplant.
Rituximab-CHOP is commonly used in non-transplant candidates within the overall response rate of greater than 90%, but a median progression free survival of only 16-21 months (whichHoward OM et al, Lenz G et al).
In a study comparing bendamustine and rituximab with standard R-CHOP therapy in 514 patients with untreated indolent non-Hodgkin’s lymphoma or mantle cell lymphoma: at a median follow-up of 45 months, median progression free survival was 69.5 months in the bendamustine group versus 31.2 months for the R-CHOP group (Rummel MJ et al).
Bendamustine-rituximab should be considered first-line therapy for mantle cell lymphoma.
Despite high-dose chemotherapy with autologous stem cell transplantation or high-dose chemotherapy (hyperCVAD) late relapses still occur in most patients.
Bortezomib presently (2010) only drug approved for relapsed disease with a response rate of 33%, lasting for a number of months (Fisher RI).
Response rate with bortezomib 42%.
Bendamustine plus rituximab versus rituximab-CHOP had a 92.7% versus 91.3% overall response rate, respectively and the progression free survival was 33 months versus 23 months, respectively (Rummel MJ et al).
R-CHOP followed by rituximab maintenance therapy is presently the standard of care for elderly patients with MCL ( European Mantle Cell Lymphoma Network)
4 courses of hyperCVAD (hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone) alternating with high dose methotrexate and cytarabine and consolidated with high dose chemotherapy and allogeneic stem cell transplantation results in an overall response rate of 93% (and 100% of all transplanted patients), with a 3-year event free survival and overall survival of 72% and 92%, respectively. By 5 years the disease free survival was 43% and overall survival 77%.
The addition of rituximab to HyperCVAD demonstrates an 87% complete response rate and a 40 month overall survival rate of 81%.
No standard therapy exists, but with advanced disease cytotoreductive therapy with rituximab based treatment, followed by high-dose therapy or autologous stem cell transplant in patients in remission is advocated.
Responds to many different chemotherapy treatments, although the duration of response is short and subsequent regimens have even shorter and shorter durations of response with drug resistance emerging.
Nonmyeloablative allogeneic stem cell transplantation may be associated with a five year, progression free survival as high as 51% (Sorror ML).
Patients who receive maintenance rituximab following autologous stem cell transplant have significantly better overall survival, compared with patients who were followed with observation alone after transplant.
Temsirolimus, any inhibitor of mammalian target of rapamycin, has shown a response rate of 38% (Witzig TF).
The global phase II trial with Ibrutinib 560 mg daily until progression or toxicity in recurrent MCL, showed an ORR of 68% with a 21% CR and a median duration of of response 17.5 months.
Rituximab in combination with lenalidomide have produced a response rate of 53% in relapse MCL (Wang L).
Lenalidomide has an overall response rate of 26% and a median duration of response of 16.6 months in patients with relapsed or refractory MCL.
A combination of Lenalidomide and rituximab results in a response rate of 92%, a complete response rate of 64%, with a medium progression free survival that had not been reached as initial treatment.
The two-year progression free survival with a combination of lenalidomide and rituximab was 85% and the 2 year overall survive was 97% (Arian J et al).
SAR3419, an anti-CD19 antibody immunogonjugate associated with a 68% response rate (Younes A).
Poor prognosis includes leukemic phase, bone marrow involvement, high mitotic rates, poor performance status and B symptoms.
Maintenance therapy with ibrtutinib and rituximab are considered choices.
Some patients with mantle cell lymphoma do not require immediate therapy and can be safely observed:This includes patients with nodal presentation but with low volume disease and no symptoms.
Patients who require therapy are divided into those patients who are young and healthy and candidates for aggressive therapy and another group that are poor candidates for such.
Chemotherapy/immunotherapy agents are administered before the use of autologous bone marrow transplantation.
Rituximab maintenance is a key factor influencing survival in patients with mantle cell lymphoma (MCL) who had undergone autologous stem cell transplant (ASCT).
Combination therapy with bendamustine and rituximab-and R-CHOP currently first line therapies.
Maintenance therapy with bendamustine and rituximab is beneficial for maintaining remission and can be considered for up to 2 to 3 years.
Acalabrutinib (Calquence) highly efficacious treatment.
Acalbrutinib had a 80% overall response rate with a 40% complete response rate.
Lenalidomide plus rituximab associated with a a high raate of response.
Ibrutinib associated with a 26% complete remission rate and at 3 years 1/4 of patients have not progressed.
Ibrutinb plus bendamustine and rituximab leads to a strong progressive free survival advantage without an overall survival advantage.
A combination of rituximab, lenalidomide and ibrutinib associated with a response rate over 85% and a complete remission rate of 60% in relapsed or refractory patients.
Rituximab maintenance after combination chemotherapy leads to a better progression free survival and overall survival.
Dual targeting therapy with Bruton’s tyrosine kinase inhibitor ibrutinib and venetoclax a BCL2 inhibitor associated with a high response rate.
A CAR T-cell therapy ((brexucabtagene autoleucel)) as a treatment for adult patients with relapsed/refractory mantle cell lymphoma.
In most patients with mantle cell lymphoma, the primary treatment regimen does not result in a cure and salvage therapy is required.
In salvage therapy mantle cell lymphoma is one of the most radiosensitive of all non-Hodgkin’s lymphomas, and the response rated of any treated site even to low dose radiotherapy, is higher than the rate could be expected with further chemotherapy treatments.
In this setting symptom relief can be achieved in more than 90% of patients with a response rates in sites treated by radiotherapy ranging from 85 to 100%, and complete remission ranging from 60 to 70%.
In the rare case of localized relapse, radiotherapy should be considered.
However, for most patients with mantle cell lymphoma, systemic disease and long-term control requires systemic therapy.
In salvage therapy, regimens with traditional chemotherapy, with the without rituximab or bendamustine and fludarabine have been highly active.
Combination of rituximab, dexamethasone, cytarabine and platinum followed by autologous stem cell transplant and maintenance rituximab was associated with event free survival rate for years is 79%.
Targeted therapies are active in patients with relapsed or refractory mantle cell lymphoma and include: Bruton’s tyrosine, kinase inhibitors Ibrutinib, Zanbrutinib, acalabrutinib, and lenalidomide , bortezomib, and m-TOR inhibitors gem sirolimus, and everolimus, PIK3 inhibitors idealisib, and umbralisib and BCL2 inhibitor venetoclax.
The combination of Ibrutinib and Venetoclax- complete response rate was 62-71% in relapsed patients.
Autologous hematopoetic stem cell transplant for relapse or refractory disease has a very high relapse rate of 80 to 90% at five years.
Allogeneic and hematopoetic transplants have high treatment related death rate and morbidity , but long remissions and probable cure in 30 to 50% of treated patients.
Checkpoint inhibitors on minimally active.
Bispecific, T cell engagers and chimeric antigen receptor T cell therapy appear advantageous.