 Autosomal genetic disorder that leads to progressive loss of cognitive, motor, and emotional function and eventually results in death.
Autosomal genetic disorder that leads to progressive loss of cognitive, motor, and emotional function and eventually results in death.
A neurodegenerative genetic disorder that affects muscle coordination and leads to cognitive decline and psychiatric problems.
Most common genetic cause of abnormal involuntary writhing movements called chorea.
Almost all patients ultimately exhibit similar physical symptoms, but the onset, progression and extent of cognitive and psychiatric symptoms vary significantly between individuals.
The most characteristic initial physical symptoms are jerky, random, and uncontrollable movements, chorea.
Chorea may be initially present as restlessness, small unintentional, uncompleted motions, lack of coordination, or slowed saccadic eye movements.
More common in people of Western European descent than in those of Asian or African ancestry.
The disease is caused by an autosomal dominant mutation in either of an individual’s two copies of a gene called Huntingtin, which means any child of an affected person typically has a 50% chance of inheriting the disease.
Symptoms can begin at any age from infancy to old age, but usually begin between 35 and 44 years of age.
The disease may develop earlier in life in each successive generation.
About 6% of cases start before the age of 21 with an akinetic-rigid syndrome or Westphal variant HD.
More than 15,000 Americans have the disease and at least 150,000 others have a 50 percent risk of developing the disease.
Symptoms vary between individuals and even among affected members of the same family, but usually progress predictably.
Symptoms appear in middle age.
Early symptoms include uncontrolled movements, clumsiness or balance problems.
Eventually may impair ability to walk, talk or swallow.
The earliest symptoms are often subtle problems with mood or cognition.
Minor motor abnormalities usually precede more obvious motor dysfunction by at least three years.
Rigidity, writhing motions or abnormal posturing appear as the disorder progresses.
Psychomotor functions become increasingly abnormal with impaired stability, altered facial expression, difficulties chewing, swallowing and speaking.
Sleep disturbances may occur.
Juvenile disease generally progresses faster and chorea is exhibited briefly, if at all, with rigidity being the dominant symptom.
Seizures are also a common symptom of this form of disease.
Behavioral and psychiatric symptoms are common and include: Irritability 38–73%, apathy 34–76%, anxiety 34–61%, depression 33–69%, obsessive and compulsive behavior 10–52%, and psychosis 3–11%.
Cognitive functions are impaired as the disease progresses.
With progression of the disease process memory deficits tend to appear.
Cognitive problems Increase over time, ultimately resultming in dementia.
Eating difficulties commonly cause weight loss and malnutrition.
Lack of coordination and an unsteady gait often follows. and with progression uncoordinated, jerky body movements become more apparent, along with a decline in mental abilities and behavioral and psychiatric problems.
Coordinated movement becomes very difficult, and mental abilities decline into dementia.
Secondary complications of the process reduce life expectancy to around twenty years after symptoms begin.
No cure for the disease.
Currently, treatment is limited to therapies that treat symptoms.
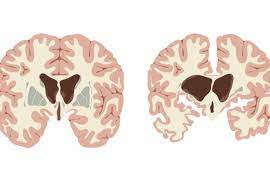
Affected are cells of the basal ganglia, especially targets neurons of the striatum, particularly those in the caudate nuclei and the pallidum.
The striatum’s subthalamic nuclei send control signals to the globus pallidus.
The globus pallidus initiates and modulates motion.
Also affected is the brain cortex, which controls thought, perception, and memory.
Results in subcortical dementia syndrome to distinguish it from the typical effects of cortical dementias such as Alzheimer’s disease.
Neuropsychiatric manifestations include anxiety, depression, blunted affect, egocentrism, aggression, and compulsive behavior.
May worsen addictions to alcohol, gambling, and hypersexuality.
Llifetime prevalence of psychiatric disorders between 33% and 76%.
Suicidal ideation and suicide attempts are more common than in the general population.
As mutant Huntingtin is expressed throughout the body and is associated with abnormalities in peripheral tissues and include muscle atrophy, cardiac failure, impaired glucose tolerance, weight loss, osteoporosis and testicular atrophy.
People have two copies of the Huntingtin gene (HTT, HD) which codes for the protein Huntingtin (Htt).
Part of the gene is a repeated section called a trinucleotide repeat, which varies in length between individuals and may change length between generations.
When the length of this repeated section reaches a certain threshold, it produces an altered form of the protein, called mutant Huntingtin protein (mHtt).
The Huntington’s disease mutation is genetically dominant and almost fully penetrant.
The Huntingtin mutation of either of a person’s genes causes the disease.
The abnormal gene results in the production of mutant HTT, containing an expanded polyglutamine tract, which causes neuronal dysfunction and death by means of toxic gain-of-function mechanisms.
In Huntington’s disease, a genetic mutation occurs in the HTT gene which encodes for Htt protein.
The Htt protein interacts with over 100 other proteins, and appears to have multiple biological functions.
The behavior of this mutated protein is not is toxic to certain cell types, particularly in the brain, and is most evident in the striatum, but as the disease progresses, other areas of the brain are also more conspicuously affected.
Early symptoms are attributable to functions of the striatum and its cortical connections—namely control over movement, mood and higher cognitive function.
Huntington disease causes astrogliosis and loss of medium spiny neurons.
In Huntington’s chorea the areas of the brain are affected according to their structure and the types of neurons they contain.
Brain structures decrease in size as they cumulatively lose cells.
In Huntington’s chorea the areas affected are mainly in the striatum, but also the frontal and temporal cortexes.
It is not inherited according to gender.
The disease is inherited by the length of the repeated section of the gene, and the severity can be influenced by the sex of the affected parent.
People have two copies of the Huntingtin gene (HTT, HD) which codes for the protein Huntingtin (Htt).
Part of the gene is a repeated section called a trinucleotide repeat, which varies in length between individuals and may change length between generations.
When the length of this repeated section reaches a certain threshold, it produces an altered form of the protein, called mutant Huntingtin protein (mHtt).
The Huntington’s disease mutation is genetically dominant and almost fully penetrant.
The Huntingtin mutation of either of a person’s genes causes the disease.
It is not inherited according to gender.
The disease is inherited by the length of the repeated section of the gene, and the severity can be influenced by the sex of the affected parent.
The HTT gene is located on the short arm of chromosome 4[13] at 4p16.3.
HTT contains a sequence of three DNA bases—cytosine-adenine-guanine (CAG)—repeated multiple times – CAGCAGCAG-known as a trinucleotide repeat.
CAG is the genetic code for the amino acid glutamine, and a series of them results in the production of a chain of glutamine known as a polyglutamine tract, and the repeated part of the gene, the PolyQ
Disease status, depends on the number of CAG repeats.
Individuals who have fewer than 36 repeated glutamines in the polyQ region results in production of the cytoplasmic protein Huntingtin.
A sequence of 36 or more glutamines results in the production of a protein which has different characteristics and is called mHtt (mutant Htt), and it increases the degeneration rate of certain types of neurons.
The number of CAG repeats is related to how much this process is affected, and accounts for about 60% of the variation of the age of the onset of symptoms, with the remaining variation attributed to environment and other genes that modify the mechanism of HD.
36–40 repeats result in a reduced-penetrance of the disease, with a much later onset and slower progression of symptoms.
With very large repeat counts full penetrance can occur under the age of 20, and is called juvenile,, accounting for about 7% of HD .
Tthe phenotype does not skip generations.
An affected individual typically inherits one copy of the gene with an expanded trinucleotide repeat , the mutant allele, from an affected parent.
Penetrance of the mutation is very high, so those who have a mutated copy of the gene will have the disease.
Trinucleotide CAG repeats over 28 are unstable during replication and this instability causes the number of repeats to change in successive generations, such that an unaffected parent with 28–35 repeats or reduced penetrance with 36–40 repeats, may pass on a copy of the gene with an increase in the number of repeats that produces fully penetrant HD.
Rarely the onset of HD may be so late that symptoms are not recognized.
Within the basal ganglia, targets neurons of the striatum, particularly those in the caudate nuclei and the pallidum.
Also affected is the cortex, which controls thought, perception, and memory.
The Huntingtin gene provides the genetic information for a protein huntingtin.
Expansion of a CAG triplet repeat stretch within the Huntingtin gene results in a mutant form of the protein, which gradually damages cells in the brain.
Genetic testing can be performed at any stage of the disease, even before the onset of symptoms.
Caused by expansion of CAG trinucleotide repeat in exon 1 of the gene that encodes huntington protein.
If the gene for the disease contains more than 35 CAGs that code for a glutamine residue, the disease is likely to develop.
Studies reveal increased atrophy in asymptomatic patients and those with early disease compared with controls.
huntington’s
Increases in the number of repeats is associated with earlier age of onset and severity of disease in successive generations in a phenomenon known as genetic anticipation.
Genetic Instability in HD is greater in spermatogenesis than oogenesis.
Maternally inherited alleles are usually of a similar repeat length, whereas paternally inherited ones have a higher chance of increasing in length.
Huntington’s disease is rarely caused by a new mutation, where neither parent has over 36 CAG repeats.
When both parents have an expanded HD gene, the risk of developing HD increases to 75%.
When either parent has two expanded copies, the risk for the development is 100%.
It is rare for an individual to have both genes affected.
The Htt protein interacts with over 100 other proteins, withmultiple biological functions.
Mutant huntingtin protein is toxic to certain types of cells, particularly in the brain.
Early damage is most evident in the striatum of the brain.
With progression of the process other areas of the brain are affected such as control over movement, mood and cognition.
Htt is expressed in all cells, but. highest concentrations are found in the brain and testes, with lesser amounts in the liver, heart, and lungs.
Htt interacts with proteins which are involved in transcription, cell signaling and intracellular transporting, and is anti-apoptic.
Htt facilitates vesicular transport and synaptic transmission and controls neuronal gene transcription.
Expression of increased Htt improves brain cell function.
HD is not caused by inadequate production of Htt, but by a gain of toxic function of mHtt.
Process affects the whole brain, but the most prominent early effects are in a part of the basal ganglia called the neostriatum, which is composed of the caudate nucleus and putamen.
Other areas affected include the substantia nigra, layers of the cerebral cortex, the hippocampus, purkinje cells in the cerebellum, lateral tuberal nuclei of the hypothalamus and parts of the thalamus.
Neurological areas are affected according to their structure and the types of neurons they contain, reducing in size as they lose cells.
Striatal neurons are the most vulnerable.
Causes an abnormal increase in astrocytes and activation of the brain’s immune cells, microglia.
The basal ganglia is the part of the brain most prominently affected in early HD and it inhibits a large number of circuits that generate specific movements.
Damage to the basal ganglia can cause inhibitions to be erratic and uncontrolled, resulting in awkward and erratic motions poorly initiated, halted before, or beyond, their completion.
CREB-binding protein (CBP), a transcription factor, essential for cell function, acts as a coactivator at a significant number of promoters, is reduced in patients brains in HD.
Medical diagnosis can be made following the appearance of physical symptoms specific to HD, and genetic testing can be used to confirm a diagnosis if there is no family history.
Genetic testing can confirm if an individual or embryo carries an expanded copy of the trinucleotide repeat in the HTT gene that causes the disease.
A positive genetic result is not considered a diagnosis, since it may be obtained decades before the symptoms begin.
Negative genetic testing means that the individual does not carry the expanded copy of the gene and will not develop the disease,
Excessive unintentional movements of any part of the body suggest a diagnosis of HD.
Huntington’s disease rating scale provides an overall rating system based on motor, behavioral, cognitive, and functional assessments.
While medical imaging, such as computerized tomography (CT) and magnetic resonance imaging (MRI), can show atrophy of the caudate nuclei, these changes are not diagnostic.
Cerebral atrophy can be seen in the advanced stages.
Over 95% of individuals at risk of inheriting HD do not proceed with testing.
The risk of suicide is increased after a positive test result.
Genetic counseling important part of management.
It is possible to obtain a prenatal diagnosis for an embryo or fetus in the womb, using fetal genetic material acquired through chorionic villus sampling.
About 99% of HD diagnoses can be confirmed by genetic testing to have the expanded trinucleotide repeat that causes HD.
No cure for HD exists presently.
Some evidence for the usefulness of physical therapy, occupational therapy, and speech therapy to improve quality of life.
Tetrabenaze can be used for the treatment of chorea.
Additional agents to reduce cornea include neuroleptics and benzodiazepines.
Hypokinesia and rigidity can be treated with antiparkinsonian agents.
Valproic acid may be used to treat HD related myoclonic hyperkinesia.
Weight loss and eating difficulties may present nutritional problems late in the disease.
Physical therapy is beneficial to maintain motor and functional performance.
The length of the trinucleotide repeat accounts for 60% of the variation in the age symptoms appear and the rate they progress.
Life expectancy is approximately 20 years following the onset of symptoms.
Life-threatening complications result from lack of muscle coordination and behavioral changes from declining cognitive function.
Pneumonia causes death in one third of cases, as difficulty clearing the lungs, and aspiration risks increase the risk of contracting pulmonary infection.
Heart disease causes about one quarter of deaths of HD.
Suicide causes greater than 7.0% of fatalities and up to 27% attempts to do so.
Worldwide prevalence of HD is 5-10 cases per 100,000 persons.
Prevalence is similar for men and women.
Frequency is highest in Western Europe, averaging around seventy per million people, and is lower in the rest of the world.
One of the highest prevalence areas is in the isolated population of the Lake Maracaibo region of Venezuela, where the process affects up to seven thousand per million people.
Huntington’s disease in Venezuela encompasses more than 18,000 individuals spanning 10 generations.
Because of ability of the laboratory confirm the diagnosis the prevalence and incidence of the disease is increasing.
Deutetrabenazine (Austedo) approved for the treatment of chorea associated with Huntington’s disease.