 IgA-associated vasculitis, formerly called Henoch-Schönlein purpura, is a vasculitis that affects primarily small vessels.
IgA-associated vasculitis, formerly called Henoch-Schönlein purpura, is a vasculitis that affects primarily small vessels.
Immunoglobulin A–Associated Vasculitis (IgAV) (Henoch-Schönlein Purpura)
Occurs most often in children.
IgA vasculitis is the most common vasculitis of childhood.
Common manifestations include palpable purpura, arthralgias, GI symptoms and signs, and glomerulonephritis, with Hematuria and proteinuria.
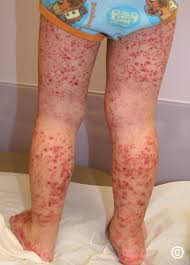
Usually begins with a sudden palpable purpuric rash typically occurring on the feet, legs, and, occasionally, the trunk and arms.
The purpura may start as urticaria that become palpable and sometimes hemorrhagic and confluent.
Diagnosis is usually a clinical one in children but usually warrants biopsy in adults.
Disease is usually self-limited in children.
Diagnostic guidelines require the presence of palpable purpura in addition to abdominal pain, arthritis or or arthralgia, renal involvement or compatible histopathological evidence.
Elevated IgA levels are neither necessary nor sufficient for diagnosis.
It is is classified as a small vessel vasculitis and most commonly affects White and Asian children, with a slight male predominance.
Disease is usually chronic in adults.
Corticosteroids can relieve arthralgias and GI symptoms but do not alter the course of the disease.
Progressive glomerulonephritis may require high-dose corticosteroids and cyclophosphamide.
IgA-containing immune complexes are deposited in small vessels of the skin and other sites.
It is preceded by infection in most patients.
The primary function of IgA is mucosal immunity, and the most common implicated organism is group A beta hemolytic strep.
Inciting antigens include viruses that cause URIs, streptococcal infection, drugs, foods, insect bites, and immunizations.
The IgA vasculitis primarily involves the skin, G.I. tract, joints, and kidneys with involvement in 95%, 70%, 70 to 90%, and 40 to 50% of cases, respectively.
Some studies suggest kidney involvement is more common and micro hematuria is present in most patients.
14% of male patients have orchitis and rarely CNS involvement is described.
Skin involvement is almost universal with IgA vasculitis.
The skin involvement reveals a petechial or purpuric rash in dependent areas typically lower legs or buttocks and other skin manifestations including bullae, edema, and the cross of the crosses can be
in infants the rash may be limited to the arms in face. Skin biopsies show leukocytoclastic vasculitis with IgG a deposition in the vessels.
Patients experience focal, segmental proliferative glomerulonephritis, but is usually but mild.
New lesions may appear over days to several weeks.
Many patients experience fever and polyarthralgia with periarticular tenderness and swelling of the ankles, knees, hips, wrists, and elbows.
GI symptoms are common and include abdominal pains and tenderness, melena.
Intussusception occasionally develops in children.
Symptoms usually remit after about 4 weeks, but often recur at least once.
Diagnosis is suspected in patients, particularly children, with typical skin findings.
Diagnostic confirmation is by biopsy of skin lesions when leukocytoclastic vasculitis with IgA in the vessel walls is identified by immunofluorescence.
IgA vasculitis can affect the G.I. tract and manifest as upper or lower G.I. bleeding.
Bowel edema can be seen causing intussusception and up to 3% of patients.
Most intussusception are iliocolic but most IgA vasculitis intussusceptionsb are ilioileal.
In the majority of cases the rash precedes the onset of G.I. symptoms.
When clinical diagnosis is clear in children, biopsy is not needed.
Urinalysis showing hematuria, proteinuria, and RBC casts is indicative of renal involvement.
Joint involvement is the second most common manifestations of IgA vasculitis.
Large joints in the legs are classically involved.
Kidney disease manifests identically to IGA nephropathy with mesangial deposits of IgA Immune complexes on histopathological examination.
Microscopic hematuria is found in 25% of patients, mild proteinuria in 15%, and 1 to 2% of patients chronic kidney disease develops.
Renal biopsy may help define the prognosis, as diffuse glomerular involvement or crescent formation in most glomeruli predicts progressive renal disease.
Most renal disease is apparent within four weeks after diagnosis and abnormal urinalysis resolves within 18 months.
The current recommendation is to monitor kidney function and blood pressure for six months after diagnosis.
Treatment: for adults, primarily symptomatic measures and corticosteroids with or without an immunosuppressant
For children, symptomatic treatment for pain control as needed, and if the cause is a drug, it has to be stopped.
For adults, corticosteroids may help control abdominal pain and are occasionally needed to treat severe joint pain or renal disease.
Pulse IV methylprednisolone followed by oral prednisone and cyclophosphamide can be given to attempt to control inflammation when the kidneys are severely affected patients.
Corticosteroids are usually not necessary for children.
Most common in children, males more commonly affected than girls with purpuric skin lesions episodic abdominal pains, arthralgias, fever, malaise, hematuria, red blood cell casts and proteinuria.
An acute small vessel leucocytoclastic vasculitis.
Most common vasculitis in children with an incidence of about 10 cases per 100,000 a year.
Median age of 6 years with 75% of patients under the age of 8 years and 90% less than 10 years of age.
Severity of the disease is milder in infants under 2 years and worse in adults Henoch-Schönlein purpura-often follows a respiratory infection and is common in autumn to spring.
A wide variety of pathogens associated with the disease and include; drugs and environmental exposures.
A minority of patients have a concomitant or recent streptococcal infection, but most cases not related to such an infection.
A positive throat culture for a beta-hemolytic streptococci reported in10-30% of patients and titers to anti-streptolysin O elevated in 20-50% of patients.
IgA1 is involved with this process with clinical manifestations secondary to vasculitis resulting from deposition into vessel walls and the renal mesangium.
IgA1 contains a hinge region with multiple O-linked glycosylation sites, but in this syndrome glycosylation is diminished, resulting in macromolecular complexes of IgA1, which when activated by complement, are deposited in the renal mesangium. Associated with mesangial hypercellularity.
Usually self-limited disorder with remissions and relapses that clear within a few months to a year.
About 10% progress to end-stage renal disease.
More than half of involved patients recover from their renal disease completely.
Older age and persistence of nephrotic syndrome imply a poorer prognosis.
Cutaneous purpura occurs in 100% of patients and is essential for the diagnosis.
Purpuric rash consist of palpable lesions 2-10 mm in diameter with scattered petechiae and ecchymoses among the lesions.
Purpuric rash most commonly on the buttocks and lower extremities, but not restricted to those sites.
Arthritis occurs in 75% of patients and usually affects the knees and ankles, with rare involvement of the upper extremity joints.
Arthritis in this syndrome is painful and can inhibit ambulation and may precede the rash by up the a week in 15-20% of patients.
Gastrointestinal symptoms of abdominal pain, nausea, vomiting and bleeding occur in 50-75% of patients.
30% of patients have grossly bloody stools but the majority have occult bleeding.
Intussusception occurs in 1-5% of cases and usually involves the ileum.
Gastrointestinal symptoms may precede onset of purpuric skin lesions by up to 2 weeks in 10-20% of patients. N About 40% of patients have renal involvement with thrombocytopenia most common finding of microscopic hematuria.
25% suffer with gross hematuria.
Proteinuria alone is rarely seen but usually accompanies hematuria in 60% of patients.
Nephritis rarely precedes purpura and can be seen weeks to months after the start of symptoms.
Renal histology similar to IgA nephropathy, and the diagnosis of H-S purpura nephritis relies on the concurrence of palpable purpura due to leukocytoclastic vasculitis with IgA in the walls of dermal capillaries.