 Cryopyrin-associated periodic syndrome (CAPS) is a group of rare, heterogeneous autoinflammatory diseases
Cryopyrin-associated periodic syndrome (CAPS) is a group of rare, heterogeneous autoinflammatory diseases
CAPS is characterized by interleukin 1β-mediated systemic inflammation and clinical symptoms involving skin, joints, central nervous system, and eyes.
CAPS encompasses three clinically overlapping autoinflammatory syndromes: familial cold autoinflammatory syndrome (FCAS), the Muckle–Wells syndrome (MWS), and neonatal-onset multisystem inflammatory disease (NOMID).
CAPS share a single genetic mutation and pathogenic pathway.
The keratoendotheliitis fugax hereditaria in which the autoinflammatory symptoms affect only the anterior segment of the eye.
It is an autosomal dominant process.
Patients may have features of more than one disorder.
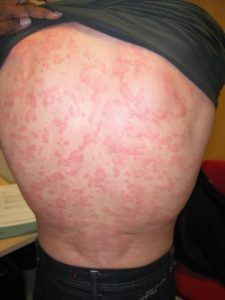
CAPS most prevalent clinical features are fever (84%), often with concurrent constitutional symptoms such as fatigue, malaise, mood disorders or failure to thrive, skin rash (97% of cases), especially after cold exposure, and musculoskeletal involvement with myalgia, arthralgia, and/or arthritis, or less commonly joint contracture, patellar overgrowth, bone deformity, bone erosion and/or osteolytic lesions (86% of cases).
Less commonly other clinical manifestations include: ophthalmological involvement with conjunctivitis and/or uveitis, optic nerve atrophy, cataract, glaucoma or impaired vision; neurosensory hearing loss (42% of cases), neurological involvement with headache, papilloedema, and/or meningitis, or less commonly seizure, hydrocephalus or mental retardation; 40% of cases), and AA amyloidosis (4%).
In keratoendotheliitis fugax hereditaria, patients experience periodical transient inflammation of the corneal endothelium and stroma, short term blurring of vision and, after repeated attacks, to central corneal stromal opacities in some patients.
Age of onset is typically in infancy or early childhood.
In 57% of cases, CAPS had a chronic phenotype with symptoms present almost daily.
43% of patients experienced only acute episodes.
Up to 56% of patients have a family history of CAPS.
CAPS are associated with a gain-of-function missense mutation in exon 3 of NLRP3, the gene encoding cryopyrin, a major component of the interleukin 1 inflammasome.
In keratoendotheliitis fugax hereditaria, the mutation occurs in exon 1.
Intracellular formation of the interleukin 1 inflammasome leads to the activation of the potent pro-inflammatory cytokines interleukin 1β and interleukin-18 through a cascade involving caspase 1.
The IL-1 inflammasome may also be released from activated macrophages, amplifying the cytokine production cascade.
NLRP3 mutations lead to aberrant formation of this inflammasome and subsequent unregulated production of interleukin 1β.
There are as many as 170 heterogenous mutations in NLRP3, inherited as an autosomal dominant with variable penetrance.
Its rarity and wide clinical presentation makes it difficult to diagnose, and a significant delay exists between symptom onset and definitive diagnosis.
Diagnosis is made by genetic testing for NLRP3 mutations.
Interleukin 1β plays a central role in the pathogenesis of the disease.
Therapy typically targets IL-1b with of monoclonal antibodies such as canakinumab, binding proteins/traps , or interleukin 1 receptor antagonists (anakinra).
These therapies are generally effective in alleviating symptoms and substantially reducing levels of inflammatory indices. Case reports suggest that
Thalidomide and the anti-IL-6 receptor antibody tocilizumab may also be effective.