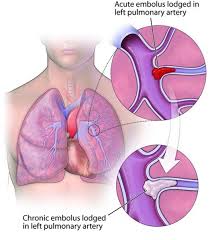
 Progressive process which is debilitating and caused by abnormally resolved thromboembolic material in the pulmonary vasculature.
Progressive process which is debilitating and caused by abnormally resolved thromboembolic material in the pulmonary vasculature.
CTEPH is characterized as primary hypertension and caused by organized thromboembolism that obstruct pulmonary blood flow and cause precapillary microvasculopathy.
The natural history of pulmonary embolism includes restoration of normal hemodynamics and gas exchange and total resolution of thromboemboli or resolution with minimal residua within 30 days.
Up to 50% of patients experience residual defects of pulmonary embolism for at least 11 months after may be at risk for developing chronic thromboembolic pulmonary hypertension.
Defined as mean pulmonary artery pressure greater than 25 mm Hg that persists 6 months after pulmonary embolism is diagnosed.
Occurs in 2-4% of patients after acute pulmonary embolism.
Up to 4% of people who suffer a pulmonary embolism go on to develop chronic thromboembolic disease including pulmonary hypertension.
At least 25% of patients with CTEPH have no prior history of pulmonary embolism.
Worldwide annual incidence estimated at five per million individuals.
Its prevalence is approximately 38 million people worldwide and is likely underdiagnosed.
True incidence is unknown because acute emboli events often occur without symptoms, and even symptomatic pulmonary embolism is overlooked or misdiagnosed.
Frequency among patients with pulmonary hypertension is unknown.
Generally present in their 40s.
Diagnosis often overlooked because of a lack of a history of a history of acute pulmonary embolism.
5-year survival rate of 10-30%.
If untreated, CTEPH typically causes premature death in greater than 50% of patients within five years of diagnosis.
The median time from symptoms to onset of diagnosis is 14 minutes.
Most patients have advanced stage, heart, failure at diagnosis.
Symptoms include shortness of breath (99%),right sided heart failure, peripheral edema (40.5%), fatigue (31.5%), chest pain (15.3%), and syncope (13.7%).
Death usually due to progressive pulmonary hypertension and right heart failure.
Increased risk for thrombophilia and genetic predisposition.
Risk factors for CTEPH include: malignancy, antiphospholipid syndrome, elevated factor VIII level and Von Willebrand factor, chronic inflammation, such as with inflammatory bowel disease, hypothyroidism, splenectomy , blood group other than type O, piece, anticoagulant, hereditary, dysfibrinogenemia, ventriculoatrial shunt, infected intravenous catheters or devices, and chronic inflammatory diseases.
Thyroid disease is a risk factor.
Diagnosis is usually made following ventilation perfusion radionuclide scanning or helical CT scans of the chest with measurement of pulmonary hemodynamics.
The screening detection on lung ventilation/perfusion scans has a sensitivity for diagnosis of 97.4% and the normal result rules out the diagnosis.
Echocardiograms are useful for assessing right ventricular dilation and estimating pulmonary artery pressure by measuring peak tricuspid regurgitation velocity, and right atrial pressure.
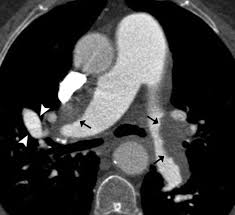
CT angiogram of the chest confirms the diagnosis and informs about location, and extent of organized thromboembolism, and has a sensitivity of 97 to 100% and a specificity of 95 to 100% for detection of thromboembolic disease in lobar pulmonary arteries, and sensitivity of 85% to 100% and specificity of 93 to 99% for detection of thrombi in segmental pulmonary arteries.
Right heart catheterization provides an accurate measurement of mean pulmonary artery pressure, pulmonary capillary wedge, pressure, pulmonary vascular resistance, and cardiac output, providing important information about prognosis and risks
All patients should be evaluated with a V/Q scan with a sensitivity is 97% in a specificity of 91% and negative predictive value of 98% for pulmonary embolism.
Severity of the disease measured by echocardiography, and pulmonary angiography.
All patients with pulmonary hypertension should undergo right heart catheterization, to evaluate mean pulmonary artery pressure before initiating therapy.
Treatment for pulmonary hypertension is pulmonary thromboendarterectomy with a mortality rate of 4.4%.
Pulmonarythromboendarterectomy involves a medium sternotomy, cardiopulmonary bypass, and brief periods of circulatory arrest to remove organized thrombus from affected pulmonary arteries.
In expert sites pulmonarythromboendarterectomy leads to substantial improvement in long-term pulmonary hemodynamics and functional status, with a perioperative mortality of less than 2 to 4%, and a five-year survival of 82%.
Approximately 25% of newly diagnosed CTEPH patients do not undergo pulmonarythromboendarterectomy due to the distal location of the lesions, and comorbidities.
Pulmonary endarterectomy requires circulatory arrest with ischemia for up to 20 minutes, as a totally bloodless field is required to perform the procedure, otherwise, the procedure will be inadequate or perforation of the pulmonary artery may occur.
Pulmonary thromboendarterectomy is a surgical procedure involving mediansternotomy and cardiopulmonary bypass to remove and organize thrombi from the pulmonary arteries.
After pulmonary thromboendarterectomy more than 95% of patients have substantial improvement in function and cardiac index and decreased pulmonary arterial pressure pressure:at one-year survival is 95%
Presently no drug therapy can prevent the process, except for anticoagulation with or without fibrinolysis treatment.
Up to 1/3 of patients with CTEPH are not candidates for the above procedure due to inaccessible thrombi or comorbidities.
Pulmonary angioplasty is considered in such patients.
Fibrinolytic treatment in hemodynamically stable patients with acute pulmonary embolism and right ventricular dysfunction can decrease the frequency of chronic thromboembolic pulmonary hypertension (Kline JA et al).
Riocigut, a soluble oral guanylate cyclase stimulator is approved for a CTEPH and improves functional capacity, decreases pulmonary vascular resistance in patients with inoperable CTEPH or recurrent pulmonary hypertension.
All patients with CTPH should receive lifelong anticoagulation, if not contraindicated.
Symptomatic residual pulmonary hypertension is estimated at 5 to 35%, in patients with higher initial pulmonary vascular resistance