 The choroid plexus (CP) of the blood-cerebrospinal fluid barrier (BCSFB) impacts CSF homeostasis, brain diseases, and neuromedical translation.
The choroid plexus (CP) of the blood-cerebrospinal fluid barrier (BCSFB) impacts CSF homeostasis, brain diseases, and neuromedical translation.
CP executes neuroendocrine, excretory, and neuroimmune actions.
BCSFB manages the brain-spinal cord fluid environments.
BCSFB tight junctions and transcellular mechanisms differ from the blood-brain barrier (BBB) counterparts, regulating pathogen and leukocyte access to the CSF-brain nexus.
Neuronal networks require stable extracellular fluid (ECF).
Neurotransmission and electrical signaling depend on ECF.
Transport/barrier/secretory systems at the blood-brain barrier (BBB) and blood-cerebrospinal fluid barrier (BCSFB) primarily regulate ECF.
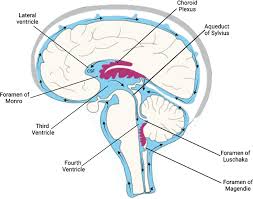
The Choroid plexus (CP) is the main locus of the BCSFB, but circumventricular organs/meninges also contribute.
BCSFB and BBB coordinate, and both barriers contain elements of the innate immune system and provide cytokine signals into brain and CSF.
The BBB and BCSFB provide substrates via regulated carrier transports to the CNS, and both act as barriers to thwart diffusion of potentially injurious molecules.
Choroid epithelial cells are specialized for transporting hormones, growth factors and neurotrophins into CSF for delivery to brain.
The BCSFB barrier also secretes vitamin micronutrients and brain-modulating proteins: transthyretin and cystatin C protease inhibitor, which enter the ventricular CSF, flowing by volume transmission to spinal cord and brain, including Virchow-Robin spaces in cortical subarachnoid space.
Leukocytes and cytokines transfer into the CSF at accelerated rate in disease, traumatic injury and infections.
Reabsorptive transporters for K and organic anions remove excesses from ECF capillaries and CSF.
The blood brain barrier and blood of the cerebrospinal fluid barrier
work in concert to maintain ECF stability in health, and restore homeostatic balance following CNS insults.
Brain microvessels chiefly translocate amino and fatty acids as well as glucose into the parenchyma .
CP secretes numerous trophic/stabilizing proteins: brain-derived neurotrophic factor. transthyretin, hormones and micronutrient vitamins.
Choroidal and cerebral endothelial secretions physically combine, especially near brain parenchyma, to form the ECF milieu for neurons.
CSF movement thoroughly mixes all fluids in the CNS.
CSF contacts virtually every barrier interface.
CP primarily generates CSF that flows down the ventricles to the basal cisterns in the subarachnoid space, from there, CSF distributes widely to far reaches of the brain/cord.
Transport activities and CSF flow create an ECF blend of BBB and CP secretions.
Fluid blending is facilitated by the perivascular system that starts as subarachnoid space CSF entering the superficial cortex at the Virchow-Robin spaces.
The driving force that propels CSF forward into the ventricular, subarachnoid and perivascular systems originates largely at the briskly-perfused and secreting CP villi.
CSF formed by CP cells flows down the ventricular system into subarachnoid space surrounding cortex, cerebellum, brain stem and spinal cord.
Ependymal cells line the ventricle walls, providing a permeable interface between CSF and brain ECF.
CSF oscillates gently to-and-fro with cardio-respiratory rhythms while undergoing bulk flow to distal reaches of subarachnoid space.
CSF under pressure flows over the cortices to drain across arachnoid villi (AV) into venous blood, or to enter the brain’s paravascular system at the Virchow-Robin spaces (VRS).
Each CP villus contains an inner vascular core, and adjacent interstitium, surrounded by a ring of epithelial cells in adults.
The choroid plexus has abundant endoplasmic reticulum, golgi apparatus and mitochondria enable profuse synthesis of proteins and peptides.
Choroidally-derived proteins/peptides secreted then flow through the ventriculo-interstitium to neuronal and glial targets.
BCSFB secretions into CSF-ECF abet healing of damaged neurons.
CP tissues have multiple villi that project like a fishnet into ventricular CSF.
Villi receive blood flow from choroidal arteries.
Plasma filters across highly-permeable capillary walls to the interstitial fluid (ISF) compartment, which is about 15% of tissue volume.
The interstitial fluid contains a matrix where cytokines/leukocytes reside, and it interacts with the basal (ISF-touching) epithelial plasma membrane.
The innermost capillaries have fenestrated permeable endothelial membrane, and leaky inter-endothelial junctions, with their
underlying basement membrane abutting the extracellular matrix.
Solutes traffic across epithelial cells is active and dynamic.
There is inward movement of plasma solutes occuring transcellularly across epithelial cells, and outward movement of CSF solutes transcellularly.
The first sequential casualty of barrier injuries is the nearby ECF composition.
Distorted ECF biochemistry disables neuronal/synaptic functions.
Active Na distribution across membranes determines the fluid balancing between extracellular and intracellular compartments.
Na transporters couple to H2O movement.
Na pump (Na-K-ATPase) faces the CNS side of the barrier and establishes steep Na gradients across the low-Na barrier cells.
Na pumping is critical for cell volume, voltage gradients, and the net trans-barrier movement of solutes.
Cerebral and CP edema as with stroke, trauma and hyperthermia is that fluid retention stems from compromised Na transport and reduced water removal.
Conversely, Na-H2O fluid movements at the barriers may increase in certain inflammatory states.
When the CNS is insulted by injuries, the CSF/plasma ratios for solutes deviate from normal.
Na transport to maintain central fluid dynamics and gradients depends on mitochondrial ATP availability.
CP epithelium and brain endothelium abound in mitochondria that ATP energizes active transport to set up the transmembrane Na gradients that drive CSF formation.
Mitochondrial failure, primary by gene mutation13 or secondary to amyloid-beta in CP as in Alzheimer’s disease, curtails CSF dynamics and disrupts solute homeostasis/gradients; cell energy failure from ATP diminution also enhances oxidative stress.
Mitochondrial apoptosis, associated with disordered genes increases in barrier injury and autoimmune disease.
Systemic inflammation also threatens the wellbeing of BBB-BCSFB.
Barriers are impaired by vascular disorders of stroke, traumatic brain injury, infections and byproducts of neurodegenerative disease, such as amyloid plaque.
Disrupted transport interfaces provokes biochemical instability and immune-brain imbalance in parenchyma of cortical, CSF and periventricular regions.
Lipopolysaccharide (LPS), disrupts the BBB and BCSFB upon penetrating the damaged barriers, LPS drives the NF-kB associated progressive inflammatory signaling in the parenchyma: a prominent feature of neurodegeneration.
Perturbed immune-brain interaction is related to many neurological/psychiatric disorders: status epilepticus, stroke/ischemia, and the neurodegenerative Parkinson’s and Alzheimer’s diseases.
BBB and BCSFB in injury states compensatorily secrete peptides, growth factors and neurotrophins thst stabiliz extracellular-intracellular fluid balance for neural health.
Several brain disorders are caused/exacerbated by disrupted barriers that encompass neuronal networks.
Barrier breaching enables central invasion by foreign substances, microbial agents and immune elements.
Consequently, ECF composition is tainted by the centrally-invading plasma proteins and immune elements normally screened by barriers.
Sentinel-like detection and resolution of neuroinflammation depend on an intact CP and neurovascular unit.
Cells of the BCSFB and BBB secrete neurotrophins, immunoregulators, and anti-inflammatory cytokines for CSF adaptation to circulating vascular stressors.
Systemic inflammation threatens BCSFB and BBB integrity as barriers become greatly permeable with dire consequences for brain.
Peripheral inflammation shifts the ECF to a pro-inflammatory state.
This local inflammatory response of cytokine production in barrier cells, is then transmitted outward into brain ECF and CSF.
BBB injury from cytokine upregulation accompanies most brain diseases and infections.
The BCSFB also responds sensitively to circulating lipopolysaccharide.
Sepsis-associated encephalopathy (SAE)
carries high morbidity/mortality and is characterized by neuroinflammation, circulatory dysfunctions, CNS barrier breakdown and cognitive impairment.
The compromised BCSFB significantly contributes to the consequent development of damaging neuroinflammation.
Bacteria stimulate CP epithelial receptors, generating a immuno-products and vesicles for transfer into CNS.
Low-grade systemic infection prompts CP to form vesicles or exosomes, containing small RNA molecules, which are discharged into the CSF, presumably for regulating astroglial/microglial targets.
Cerebral capillaries and choroid plexuses remove from extracellular fluid excesses of various solutes.
Clearance transporters remove organic anions from brain ECF and CSF.
Neurodegeneration and neuroinflammation disrupt CNS barrier transporters, and such disruption adversely affects ECF homeostasis, disease progression and neural pharmacotherapy.
CP acts in transporting hormones from blood to hypothalamus.
These include prolactin and insulin-like growth factor 1 secretion, from plasma-to-CP-to-CSF flowing by volume transmission to hypothalamic target cells.
Major transport of folate across BCSFB is essential for proper neural tube development.
CSF folate dwindling leads to spina bifida/hydrocephalus.
Transthyretin, secreted by CP epithelium facilitates thyroid hormone transport into CSF-CNS and protects neurons by stabilizing amyloid conformation.
Aquaporin 1 (AQP1) at the BCSFB facilitates water penetration into CSF.
Certain immune phenomena: leukocyte migration, may be associated more prominently with BCSFB than BBB.
Under particular conditions, the CP gate allows leukocyte penetration to downstream periventricular regions, as occurs in multiple sclerosis relapses.
Viruses, bacteria, and fungi exploit CP as a CSF port of entry into brain.
Proteins leak from the blood into brain and CSF, and this infiltration increases in aging and neurodegeneration.
Proteins diffuse across BCSFB more extensively than BBB, due to more permeable tight junctions in CP than cortical capillaries.
In neural diseases the CSF-to-serum ratios of proteins such as albumin primarily reflect BCSFB breaching:
replace the notion that the CSF-to-serum ratio of proteins mainly assesses brain microvessel permeability.
Many polypeptides and proteins, arising from BCSFB transport into CSF modulate brain viability.
Following severely-perturbed CSF-brain homeostasis, neurotransmission falters, and cognitive and behavioral losses occur, with morbidity crises and even death.
The brain is broadly immersed in CSF, and CP forms most of the CSF
The CP floats in ventricular CSF, spreading out tmaximizes surface area for microvilli exchange.
To-and-fro movement of CSF occurs responding to cardiac and respiratory pulsations, physically displaces CP villi, allows thorough mixing of CSF for monitoring and adjustments.
BCSFB is a highly dynamic transport interface with activity intensity on par with BBB.
This notion is bolstered by findings that CP-generated net turnover of CSF greatly exceeds BBB fluid generation.
Evaluating the respective barrier roles in a range of pathologies reveals precise pharmacologic targets to reconstitute injured brain.
Strong evidence implicates CP-CSF-brain solute transport and distribution as critical for CNS ontogeny.
Malfunctions of transport systems and CSF volume distribution predispose to developmental neural diseases/disorders.
Neuropsychiatric systemic lupus erythematosus (NPSLE) is an autoimmune disease caused by leukocyte and immunoglobulin penetration into CSF going to periventricular regions that become inflamed.
Formerly, a leaky BBB was implicated in neuropsychiatric SLE onset; however, a paradigm shift in transport modeling may be emerging, suggesting a perturbed BCSFB inflames the subependymal regions.
Autoantibodies to N-methyl D-Aspartate (NMDA) receptors impair neurotransmissioin as autoencephalitis.
An imbalanced inflammasome promotes neuroinflammation-mediated cognitive decline.
Autoantibodies against NMDA receptor are in CSF.
BCSFB is more permeable than BBB, especially in neurodegeneration.
Infection-induced inflammation in early life gives rise to maldevelopmental issues such as cerebral palsy.
The leukocyte/cytokine migration across BCSFB, controlled by toll like receptor activity, is a significant aspect of the innate immune response to pathogens.
Toll like receptors (TLR) in CP consist of transcripts for receptor subtypes, promoting neuroinflammation and neuronal damage.
Specific activity of TLR2, promotes neutrophil and monocyte infiltration into the CSF and downstream arachnoid membrane.
Choroid plexus-CSF gateway for trypanosomes
Trypanosome inducers of African sleeping sickness initially and rapidly enter CSF, but not the brain , after intraperitoneal injection.
Trypanosoma brucei readily passes through the BCSFBen route to eventually infecting brain following passage via ventricular CSF flow.
This microbe accesses the Virchow-Robin spaces, that are entry loci for subarachnoid CSF flow into cerebral paravascular spaces.
Trypanosomes penetrate the CNS predominantly by initial CSF access, following which there is convective distribution into the brain.
The pathophysiology of Alzheimer’s disease is more complex than the original concept of neuronal Aβ production vs. BBB clearance.
The glymphatic system, involving astrocyte transports waste clearance; it is connected downstream to a valid network in the cervical lymphatics.
Impaired drainage of Aβ in glymphatic fluid into cervical lymph is likely a contributing factor in Alzheimer pathophysiology.
Aβ distribution within CNS relates to cholesterol homeostasis and transport at the barriers.
Imbalanced cholesterol metabolism/transport in CP-CSF may harm the brain.
APOE regulates lipid distribution between plasma and body compartments: CSF and brain.
Free cholesterol exchange across BBB and BCSFB favorably affects net Aβ extrusion from CNS.
Individuals hetero- or homozygous for the APOE4 allele have greater vulnerability for disrupted barriers.
Aβ concentration stability in CNS depends on cholesterol homeostasis.
Uptake of Evans blue-albumin complex by brain shortly after intravenous injection reliably evaluates barrier leakiness.
Steady-state CSF/serum albumin values primarily reflect BCSFB permeability.
BBB refers specifically to the cerebral microvessel wall, whereas BCSFB typically designates the CP epithelial membrane.
Eosinophilic inflammation disrupts BCSFB, increasing CSF protein, albumin, plasminogen activator, matrix metalloproteinases (MMP-9 & MMP-12), TNFα and claudin-5.
Inflammatory molecules and eosinophils flow via ventricular CSF outward to the distal meninges, and infection of the arachnoid membrane usually results.
Reduced CP perfusion impairs the BCSFB.
Hippocampal regions protected by BBB also incur great damage, by a delayed response.
When highly-metabolizing CP is deprived of O2, the BCSFB epithelial disintegrates.
Ischemia-injured epithelium recovers by 24 hr.
Stroke-induced breaching of the BCSFB opens up a portalthrough which proteins, cytokines and unneeded plasma molecules freely pass into the ventricles.
Water osmotically follows molecules leaking into CSF, resulting in ventriculomegaly and brain regions proximate to CSF are destabilized.
An inflammatory stimulus, co-existing with ischemia/hypoxia, may exacerbate stroke damage by down-regulating the anti-oxidant system.
Stroke harm to BCSFB prompts plasma trafficking of monocyte-induced macrophages.
Elevated temperature increases BCSFB-BBB the permeability.
This likely occurs due to the rupturing of tight junctions between barrier cells.
Elevated temperature untoward effects on CP-CSF include choroid cell degeneration, lateral ventriculomegaly, and periventricular neuropil destabilization.
The BBB and BCSFB damage from acute hyperthermia spread widely in CSF-brain, causing the above changes.
Post-hyperthermia repair includes supportive choroidal growth factors neurotrophins, and from ventricular infusion of brain-derived neurotrophic factor (BDNF), insulin-like growth factor-1 (IGF-1) and glial derived neurotrophic factor (GDNF).
CSF flow distributes restorative peptides throughout the ventricles and adjacent brain.
Leptin upregulates in response to abdominal fat overload.
Secreted by adipocytes, leptin targets the hypothalamus for neuroendocrine signal integration of energy metabolism and thermogenesis.
Leptin accesses the CNS by high-affinity transport at the BBB and BCSFB.
Endothelial megalin expression facilitates leptin (and insulin) transport into brain to control obesity.
Leptin’s modulation of arcuate neurons helps body weight control by feedback for upregulated adipocyte lipolysis.
Resistance to leptin as a weight-regulating hormone is in two forms: i) impaired leptin transport across BBB and/or BCSFB into the hypothalamus , and ii) reduced sensitivity of leptin receptors and diminished arcuate signaling.
Leptin resistance in pregnancy, manifesting as hyperphagia results from decreased leptin transport across barriers and from altered hypothalamic signaling.
The central appetite-enhancing activity in lactation functionally relates to prolactin modulation of arcuate neurons.
Leptin actions intertwine with carbohydrate and fat metabolism.
Hyperglycemia likely upregulates barrier leptin transporters, while hypoglycemia downregulates.
Endogenous leptin fails when impaired transporters develop in obesity.
Glucagon-like peptide improvement of barriers injured by diabetes.
Diabetes mellitus injures the BBB, increasing permeability and impairs cognition.
BBB damage plays a significant role in diabetes-dependent neurodegeneration, stroke, and especially the combined disorders.
Diabetes-altered CP ion transporter expression may affect cerebral functions by modifying CSF homeostasis/dynamics.
GLP-1 diffuses rapidly across the BBB.
GLP-1 is in a new class of agents with remediation potential for elevated intracranial pressure.
A major transporter for clearing amyloid betaAβ from CNS is low-density lipoprotein receptor-related protein.
These extrusion systems normally remove Aβ sufficiently to prevent central buildup.
Tight junctions are vulnerable to pro-inflammatory cytokines.
Intestinal-derived microbial metabolites in the circulation alter tight junctions.
Regulating barrier permeability by tight junctions and Aβ transport is a dual challenge in managing neurodegeneration.
With aging and progression of late-onset Alzheimer’s disease, the BBB transporters extrude less Aβ into blood.
Subsequently the retention of Aβ by cerebral interstitium predisposes towards Aβ plaque formation and neuroinflammation.
Vitamin D supplementation holds promise for enhancing Aβ clearance to stall Alzheimer’s progression.
Supply to the brain of several endogenous substrates: vitamin C, folate, transthyretin), transported by BCSFB but not BBB.
CSF constancy and thus brain ECF stability depend on bidirectional blood-CSF transport.
Macrophages and dendritic cells accumulate in the choroidal interstitium, prior to movement into CSF.
BCSFB tight junctions are vulnerable to disintegration by matrix metalloproteinase and other injury molecules upregulated in forebrain ischemia, hyperthermia, diabetes and helminthic meningitis.
Upon leaking into CSF, the leukocytes and microbial products distribute widely.
CSF streams through the ventricles and where the Virchow-Robin spaces around major penetrating vessels admit CSF-borne materials into perivascular spaces.
CP is both a target and a gateway for throughput of microbes and bacterial products into CSF.
Lipopolysaccharides research on the BBB and BCSFB identifies the structural components of barrier breakdown, allowing the permeating LPS to provoke parenchymal inflammation and neurodegeneration.
Neuroinflammation induced by microbes threatens neuronal well-being by bacterial or cytokine battering of CP it destabilizes the BCSFB, leading to cerebral inflammation.
With a compromised BCSFB integrity cytokines and leukocytes enter into CSF/brain.
Cytokine spreading by CSF volume transmission activates microglia, promoting altered behavior.