A chronic multisystem, inflammatory process with a relapsing and remitting course.
Associated with recurrent oral and genital ulcerations, uveitis, and multisystem involvement of the intestine, joints, neurologic and vascular systems.
The BS syndrome has a substantial effect on quality of life, adversely affecting physical and psychological functions.
A multi system vasculitis that causes oral and genital ulcers, papulopustular and nodular lesions, arthritis, uveitis, arterial aneurysms, and arterial and venous thrombosis and may involve the CNS and G.I. tract.
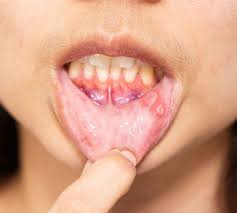
Recurrent oral alteration is the hallmark of BS, and it is the most common clinical manifestation, followed by genital ulcers, papulopustular lesions, and nodular skin lesions.
Oral ulcer lesions are both the typical and most persistent of symptoms in BS.
Up to 30% of patients have only mucous membrane manifestations throughout their illness.
It has a broad spectrum of signs and symptoms with impaired quality of life and associated morbidity, and even death.
Its pathogenesis is thought to be result of genetic predisposition after exposure to wide range of environmental triggers.
Triggers activate cellular effectors, and signaling pathways, potentially resulting in tissue inflammation and damage.
Environmental triggers include microorganisms (bacteria, viruses), consumption of histamine releasing foods (citrus, cheese), poor oral hygiene, and stress.
Evidence suggests alterations in gut and salivary mucosal flora balance that may contribute to the antigens associated with the disorder.
BS includes autoimmune and auto inflammatory features.
A systemic inflammatory disease of unknown cause.
It is classified as a primary systemic vasculitis affecting veins and arteries of any size.
Skin biopsy typically reveals leukocytoclastic vasculitis, neutrophilic, or lymphocytic perivascular infiltrates, microvascular thrombi, or neutrophilic dermal infiltrates.
The prevalence of BS in patients with HLA-B*51 is six times the general population.
Varying genes that encode endoplasmic reticulum, natural kill T cells, helper T cells, abnormal DNA methylation, and activated histones are associated with BS.
The nuclear factor KappaB, proinflammatory pathway causes immune responses in antigen presenting cells, neutrophils, and Th1 and Th17 cells.
Patients have episodic inflammation.
Neutrophils are the main infiltrating cell types in BS lesions.
Neutrophils produce excessive levels of reactive oxygen species, and release neutrophil, extracellular traps, contributing to the development of a pro coagulant state in BS.
Recurrent relapsing and remitting oral ulcers are frequently the first manifestations.
Exceedingly rare with the prevalence of approximately 0.12- 0.33 per 100,000 people.
Its current epidemiology varies according to geographic location and ethnic group.
Its highest prevalence is in Turkey with 420 cases per 100,000 persons.
In Europe the prevalence ranges from 0.3 to 4.9 cases per hundred thousand in northern countries and 1.5 to 15.9 cases per hundred thousand in southern regions.
In the US the prevalence of Behcet’s syndrome (BS) is 5.2 cases for 100,000 persons.
Familial association with BS has been reported, particularly among patients with the early onset syndrome.
The mean age at diagnosis is around 30 years.
The majority of patient present with BS present between the ages of 15 and 45 years.
Disease, activity wanes with advancing age.
There is no set difference in prevalence.
Male patient some more likely than female patients to have severe forms of the disease.
Recurrent oral ulcerations of at least three times in previous year and at least two of the following: recurrent genital ulceration, eye lesions including retinitis, uveitis, skin lesions such as erythema nodosum, or papulopustular lesions or a positive pathergy.
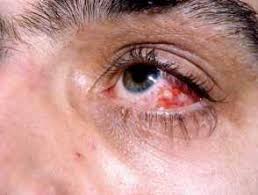
The eye is the most frequently affected major organ in BS, with ocular involvement occurring in 50% of patients.
Involvement is bilateral in 75 to 80% of new cases, on average occurring two years after the onset of initial symptoms.
Panuveitis is the most common presentation.
Anterior uveitis may lead to hypopyon,and encouraging about 10% of patients.
BS can affect both veins and arteries and typically follows a relapsing course with a five-year cumulative risk of recurrent vascular events at close to 40%.
Genital ulcers may form scars in approximately 2/3 of cases.
These also are larger, deeper and longer lasting than oral ulcers, and most appear on the scrotum or labia.
Up to 62% of patients may have erythema nodosum at presentation.
Acne-like lesions can be present indistinguishable from acne vulgaris
Associated with thrombosis at atypical sites: portal or suprahepatic veins, the superior and inferior, vena cava, and cerebral sinuses.
Sperficial thrombophlebitis, and DVT are the most common manifestations of vascular BS.
Arterial manifestations are primarily present with aneurysms, but may be present with thrombotic occlusion or stenosis of the aorta and peripheral arteries affected.
Rarely pulmonary artery involvement is present, but is highly specific for BS.
5% of patients have cardiac involvement manifesting mainly as valvular disease, but may be associated with pericarditis, myocarditis, coronary arteritis, valvulitis, cardiac thrombosis, and endocardial fibrosis.
Thrombi adhere to vein walls, and is it considered hallmark of inflammatory induced thrombosis.
An estimated 10 – 25% of patients will form thromboses and more than 27% will have thromboses as an initial sign of disease.
Vascular involvement is a significant factor in morbidity and mortality.
Approximately 50% of patients would BS have joint involvement, typically manifesting as self limited monoarthralgia, or oligo arthralgia or arthritis, primarily affecting the knees, ankles, wrists, and elbows.
Pathergy reaction refers to an exaggerated inflammatory response provoking pustule formation at the site of a needle prick.
An auto inflammatory syndrome.
Patients may have GI ulcers, seronegative arthritis, central nervous system abnormalities, skin lesions and thrombophlebitis.
Associated with recurrent oral apthous ulcers, genital ulcers and uveitis.
The main feature of Behçet’s disease is aphthous-like ulceration, that is usually more severe than seen in aphthous stomatitis without a systemic cause.
Retinal vasculitis is characteristic.
The aphthous ulceration typically resembles a herpetiforme ulceration.
Aphthous-like ulceration is the first sign of the disease in 25–75% of cases.
Papulopustular skin lesions found it up to 83% of patients at presentation and mucocutaneous ulcerations, which is considered a hallmark of disease, and other cutaneous manifestations may be seen.
More common in individuals whose ethnic origin is from regions between the Mediterranean and the Far East.
Autoimmune etiology suggested.
Associated with pathergy.
Onset of symptoms usually occurs in early adulthood.
Oral cavity agents including herpes simplex virus, Streptococcus species, Staphylococcus species and Escerichia coli proposed infectious agents that may trigger ancross reative immune response.
Ocular lesions, erythema nodosum, pustular lesion, cutaneous pathergy and bizarre neurologic findings may occur.
Vascular involvement accounts for significant morbidity.
Heat shock protein 65 found in high concentrations in oral ulcerations and skin lesions of patients with Behcet’s disease.
Heat shock proteins can stimulate the production of antibodies that cross react with streptococcal spcies in the mouth.
The underlying pathological process is a vasculitis and/or an acute neutrophilic inflammation.
Increased production of reactive oxygen species, nitrogen oxide, and auto antibodies against mucous membrane antigens play a role in the pathogenesis.
Microthromboses have been implicated in mucous membrane injury.
Bone marrow dysplasia is frequent.
The BS lacks pathopneumonic biologic or histologically diagnostic features.
The prevalence of HLA-B51 carries state varies by ethnic group.
Diagnosis is largely based on patient’s clinical presentation and imaging findings, because of the broad spectrum of disorders in the differential diagnosis.
Clinical indicators that suggest BS include genital scarring, and distinctive eye or major vascular involvement, as well as neurologic lesions from the basal ganglia to the brainstem.
Laboratory findings demonstrate inflammation with increased acute phase reactant response.
Treatment:
The overall goal is the prompt control of inflammation to prevent irreversible organ damage and relapses.
Goals of treatment include reducing the number, duration and frequency of mucocutaneous lesions, reducing joint pain and swelling, controlling ocular inflammation, ensuring visual acuity, control of vascular inflammation and prevention of new vascular, lesions, managing the post thrombotic syndrome and controlling neurologic inflammation, preventing new nervous system inflammatory lesions, preventing thrombosis and neurologic sequelae.
Immunosuppressive agents are the treatment of choice.
Patients respond well to prednisone and biological agents such as infliximab.
Colchicine is recommended as first line treatment for skin and mucosal involvement, but does not show efficacy for oral ulcer treatment.
Colchicine is used to prevent recurrence of mucocutaneous and joint lesions.
In patients with oral ulcers with Behcet’s syndrome Apremilest resulted in a greater reduction in the number of oral ulcers than placebo, but was associated with adverse effects including diarrhea, nausea, and headache.
Topical glucocorticoids, anti-inflammatory mouthwashes, and intra-articular glucocorticoid injections are also used for recurrent disease manifestations.
Ustekinumab, an inhibitor of Interleukin 12 and interleukin 23 has demonstrated efficacy in treating oral ulcers refractory to colchicine.
The prognosis depends on major organ involvement, and is predictive of both illness severity and death.
Patient with large burden of disease are most likely to have severe manifestations.
Male gender is associated with increased risk of death.
Other hazards associated with death in Behcet syndrome, are arterial involvement, high frequency of flares.
The ocular syndrome with loss of vision, occurs in approximately 25% of cases over a 10 year period: with newer therapies the risk has reduced to about 13%.
Ocular involvement warrants treatment with glucocorticoids and systemic immunosuppressants.
Neurologic BS can lead disability or death.
The risk of severe disability or death ranges from approximately 25% at seven years to as high as 60% of 10 years.
The leading cause of death in BS is the vascular syndrome, mainly due to arterial aneurysms of the aortic, pulmonary arteries and the Budd-Chiari syndrome.
Overall mortality is estimated at 5% over 7.7 years and 9.8% at 20 years.
For vascular and neurologic involvement, combinations of glucocorticoids and immunosuppressive agents are typically used.
For major organ involvement treatments include immunosuppressive agent, such as cyclophosphamide and a TNF inhibitor.