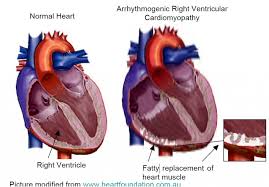
 Arrhythmogenic cardiomyopathy (AC) is a group of heart muscle disorders characterized by the replacement of normal heart muscle with fibrous and fatty tissue, primarily affecting the right ventricle, though it can also involve the left ventricle.
Arrhythmogenic cardiomyopathy (AC) is a group of heart muscle disorders characterized by the replacement of normal heart muscle with fibrous and fatty tissue, primarily affecting the right ventricle, though it can also involve the left ventricle.
Other names Arrhythmogenic right ventricular cardiomyopathy (ARVC), arrhythmogenic right ventricular dysplasia (ARVD).
The prevalence of ACM is about 1/10,000 in the general population in the United States, although some studies have suggested that it may be as common as 1/1,000.
Key features of arrhythmogenic cardiomyopathy include: replacement of heart muscle cells with fibrous and fatty tissue, which disrupts normal electrical signals and weakens the heart muscle
It is often hereditary, with mutations in genes encoding cardiac desmosomes .
Palpitations, syncope, chest pain, and in some cases, sudden cardiac death, particularly during physical exertion may occur.
Diagnosis: involves electrocardiogram (ECG), cardiac imaging (echocardiography, MRI), genetic testing, and possibly electrophysiological studies
Treatment may include antiarrhythmic medications, implantable cardioverter-defibrillators (ICDs), radiofrequency ablation, and lifestyle modifications, especially limiting strenuous exercise.
This condition is a significant cause of sudden cardiac death in young people and athletes.
Arrhythmogenic cardiomyopathy (ACM) is an inherited heart disease.
Its micro-histologic features are of ongoing myocyte death with early fibrosis and adipocyte infiltration.
ACM is caused by genetic defects of parts of the cardiac muscle known as desmosomes, areas on the surface of muscle cells which link them together.
Desmosomes are composed of several proteins, which can harmful mutations.
ARVC can also develop in intense endurance athletes in the absence of desmosomal abnormalities.
Exercise-induced ARVC is possibly a result of excessive right ventricular wall stress during high intensity exercise.
This disease is a type of non-ischemic cardiomyopathy that primarily involves the right ventricle.
It is characterized by hypokinetic areas involving the free wall of the ventricle, with fibrofatty replacement of the myocardium, with associated arrhythmias often originating in the right ventricle.
ACM can be found in association with diffuse palmoplantar keratoderma, and woolly hair, in an autosomal recessive condition, Naxos disease, because this genetic abnormality can also affect the integrity of the superficial layers of the skin most exposed to pressure stress.
ACM is an important cause of ventricular arrhythmias in children and young adults.
It is seen predominantly in males, and 30–50% of cases have a familial distribution.
Patients may not have any symptoms at all despite having significant abnormalities in the structure of their hearts.
The initial presentation is often due to arrhythmias which in arrhythmogenic cardiomyopathy may take the form of palpitations, or blackouts.
Sudden death may be the first presentation of ACM without any preceding symptoms.
These symptoms often occur during adolescence and early adulthood, but signs of ACM may rarely be seen in infants.
As ACM progresses, the muscle tissue within the ventricles may dilate and weaken leading to fatigue and ankle swelling. In the later stages of the disease in which both ventricles are involved shortness of breath may develop, especially when lying flat.
ACM is usually inherited in an autosomal dominant pattern, with variable expression.
Only 30% to 50% of individuals affected by ACM will test positive to one of the known genetic mutations in chromosomal loci associated with the disease.
Point mutations in genes encoding for desmosomal proteins are the main causatives for the development of ACM.
Desmin is an intermediate filament protein, which is linked to the desmosomes.
Different DES mutations cause an abnormal aggregation of desmin and associated proteins.
Seven gene loci have been implicated in ACM.
Excessive long-term sports activity can cause exercise-induced arrhythmogenic right ventricular cardiomyopathy.
This could possibly be a result of excessive right ventricular (RV) wall stress during very high volumes of training, which is known to be causing a disproportionate remodeling of RV.
The pathogenesis of ACM is largely unknown, but apoptosis-programmed cell death appears to play a role.
Aneurysmal dilatation is present in 50% of cases at autopsy.
The left ventricle is involved in 50–67% of individuals.
If the left ventricle is involved, it is usually late in the course of disease, and confers a poor prognosis.
There are two pathological patterns: Fatty infiltration and fibro-fatty infiltration.
Fatty infiltration at first, fatty infiltration, is confined to the right ventricle.
There is partial or near-complete substitution of myocardium with fatty tissue without wall thinning, and involves predominantly the apical and infundibular regions of the RV.
No inflammatory infiltrates are seen in fatty infiltration, but there is evidence of myocyte degeneration and death seen in 50% of cases of fatty infiltration.
Fibro-fatty infiltration, involves replacement of myocytes with fibrofatty tissue.
A patchy myocarditis is involved in up to 2/3 of cases, with inflammatory infiltrates (mostly T cells) seen on microscopy.
Myocardial atrophy is due to injury and apoptosis leading to thinning of the RV free wall to < 3 mm thickness.
Myocytes are replaced with fibrofatty tissue, and the regions preferentially involved include the RV inflow tract, the RV outflow tract, and the RV apex.
LV free wall may be involved in some cases. Involvement of the ventricular septum is rare.
The areas involvement of the heart are prone to aneurysm formation.
Participants in strenuous physical activity have an earlier onset of symptoms and earlier mortality compared to other populations.
Ventricular arrhythmias due to ACM typically arise from the diseased right ventricle, with types of arrhythmia ranging from frequent premature ventricular complexes (PVCs) to ventricular tachycardia (VT) to ventricular fibrillation (VF).
Initiating factors of the ventricular arrhythmias may be due to triggered activity or reentry.
Ventricular arrhythmias are usually exercise-related, suggesting that they are sensitive to catecholamines.
The ventricular beats typically have a right axis deviation.
Multiple morphologies of ventricular tachycardia may be present.
Right ventricular outflow tract (RVOT) tachycardia is the most common VT seen in individuals with ACM.
The differential diagnosis for the ventricular tachycardia due to ACM include:
Congenital heart disease Repaired tetralogy of Fallot Ebstein’s anomaly Uhl’s anomaly Atrial septal defect Partial anomalous venous return Acquired heart disease Tricuspid valve disease Pulmonary hypertension Right ventricular infarction Bundle-branch re-entrant tachycardia Miscellaneous Pre-excited AV re-entry tachycardia Idiopathic RVOT tachycardia Sarcoidosis
Clinical tests are employed in making a diagnosis: including the electrocardiogram (EKG), echocardiography, right ventricular angiography, cardiac MRI, and genetic testing.
90% of individuals with ARVD have some EKG abnormality.
The most common EKG abnormality seen in ACM is T wave inversion in leads V1 to V3 , and is a non-specific finding.
RBBB itself is seen frequently in individuals with ACM possibly due to delayed activation of the right ventricle, rather than any intrinsic abnormality in the right bundle branch.
The epsilon wave is a terminal notch in the QRS complex and is due to slowed intraventricular conduction.
Epsilon waves are the most specific and characteristic finding in arrhythmogenic right ventricular dysplasia (ARVD).
Myocytes are replaced by fat, producing islands of viable myocytes in a sea of fat.
This causes a delay in excitation of some of the myocytes of the right ventricle, producing a small notch seen during the ST segment of the ECG.
The epsilon wave is found in about 50% of those with ACM.
Ventricular ectopy seen on EKG in the setting of ACM is typically of left bundle branch block (LBBB) morphology, with a QRS axis of −90 to +110 degrees.
The origin of the ectopic beats is usually from one of the three regions of fatty degeneration called the “triangle of dysplasia” the RV outflow tract, the RV inflow tract, and the RV apex.
Signal averaged ECG is used to detect late potentials and epsilon waves in individuals with ACM.
Echocardiography may reveal an enlarged, hypokinetic right ventricle with a paper-thin RV free wall.
The dilatation of the RV will cause dilatation of the tricuspid valve annulus, with subsequent tricuspid regurgitation.
Fatty infiltration of the RV free wall can be visible on cardiac MRI.
Cardiac MRI can visualize the extreme thinning and akinesis of the RV free wall.
Right ventricular angiography is considered the gold standard for the diagnosis of ACM: akinetic or dyskinetic bulging localized to the infundibular, apical, and subtricuspid regions of the RV.
The specificity is 90%.
Transvenous biopsy of the right ventricle can be highly specific for ACM, but it has low sensitivity.
False negative biopsies are common.
A biopsy sample that is consistent with ACM would have > 3% fat, >40% fibrous tissue, and <45% myocytes.
ACM is an autosomal dominant trait with reduced penetrance.
Approximately 40–50% of ACM patients have a mutation identified encoding components of the desmosome, which can help confirm a diagnosis of ACM.
Since ACM is an autosomal dominant trait, children of an ACM patient have a 50% chance of inheriting the disease-causing mutation.
Family-specific genetic testing can be used to differentiate between relatives who are at-risk for the disease and those who are not.
The diagnosis of ACM is based on a combination of major and minor criteria.
To make a diagnosis of ACM requires either 2 major criteria or 1 major and 2 minor criteria or 4 minor criteria.
Major criteria Right ventricular dysfunction Severe dilatation and reduction of RV ejection fraction with little or no LV impairment Localized RV aneurysms Severe segmental dilatation of the RV Tissue characterization Fibrofatty replacement of myocardium on endomyocardial biopsy Electrocardiographical abnormalities Epsilon waves in V1 – V3 Localized prolongation (>110 ms) of QRS in V1 – V3 Inverted T waves in V1 -V3 in an individual over 12 years old, in the absence of a right bundle branch block (RBBB) Ventricular tachycardia with a left bundle branch block (LBBB) morphology, with superior axis
Minor criteria
Right ventricular dysfunction Mild global RV dilatation and/or reduced ejection fraction with normal LV. Mild segmental dilatation of the RV Regional RV hypokinesis Tissue characterization Electrocardiographical abnormalities Late potentials on signal averaged EKG. Ventricular tachycardia with a left bundle branch block (LBBB) morphology, with inferior or unknown axis Frequent PVCs (> 500 PVCs / 24 hours)
The goal of management of ACM is to decrease the incidence of sudden cardiac death.
Individuals with ACM are considered at high risk for sudden cardiac death:
Young age Competitive sports activity Malignant familial history Extensive RV disease with decreased right ventricular ejection fraction. Left ventricular involvement Syncope Episode of ventricular arrhythmia Management options include pharmacological, surgical, catheter ablation, and placement of an implantable cardioverter-defibrillator.
Patients are advised to undergo lifestyle modification, including avoidance of strenuous exercise, cardiac stimulants, such as caffeine, nicotine, pseudoephedrine and alcohol.
ARVC patients, as well as gene carriers of pathogenic ARVC-associated desmosomal mutations, should not participate in competitive sports,and should be advised to limit their exercise programs to leisure-time activities and remain under clinical surveillance.
Pharmacologic management of ACM involves arrhythmia suppression and prevention of thrombus formation.
Sotalol, a beta blocker and a class III antiarrhythmic agent, is the most effective antiarrhythmic agent in ACM.
Other antiarrhythmic agents- amiodarone and conventional beta blockers are used guided by series ambulatory Holter monitoring, to show a reduction in arrhythmic events.
Angiotensin converting enzyme inhibitors (ACE Inhibitors) have not been proven to be helpful in ACM.
Individuals with decreased RV ejection fraction with dyskinetic portions of the right ventricle may benefit from long term anticoagulation to prevent thrombus formation and subsequent pulmonary embolism.
Catheter ablation may be used to treat intractable ventricular tachycardia, with a 60–90% success rate: 60% recurrence rate, with the creation of new arrhythmogenic foci.
An ICD is the most effective prevention against sudden cardiac death.
Indications for ICD placement in the setting of ACM include:
Cardiac arrest due to VT or VF Symptomatic VT Failed drug therapy Severe RV involvement with poor tolerance of VT Sudden death of immediate family member.
Due to the extreme thinning of the RV free wall, it is possible to perforate the RV during implantation, potentially causing pericardial tamponade.
The progressive nature of the disease may lead to fibro-fatty replacement of the myocardium at the site of lead placement, causing undersensing of the individual’s electrical activity and inability to pace the ventricle.
Heart transplant may be indicated if the arrhythmias associated with the disease are uncontrollable or if there is severe bi-ventricular heart failure that is not manageable with pharmacological therapy.
First degree family members of the affected individual should be screened for ACM, and screening should begin during the teenage years unless otherwise indicated.
Screening tests:
Echocardiogram EKG Signal averaged EKG Holter monitoring Cardiac MRI Exercise stress test
There is a long asymptomatic lead-time in individuals with ACM.
While this is a genetically transmitted disease, individuals in their teens may not have any characteristics of ACM on screening tests.
ACM is a progressive disease, as the right ventricle becomes more involved, leading to right ventricular failure.
The right ventricle fails before there is left ventricular dysfunction.
By the time the individual has signs of overt right ventricular failure, there will be histological involvement of the left ventricle.
Eventually, the left ventricle becomes involved, leading to bi-ventricular failure.
Signs and symptoms of left ventricular failure may become evident, including congestive heart failure, atrial fibrillation, and an increased incidence of thromboembolic events.
It is thought that in most patients, additional factors such as other genes, athletic lifestyle, exposure to certain viruses, may be required for a patient to eventually develop signs and symptoms of ACM.
It accounts for up to 17% of all sudden cardiac deaths in the young.