A genetic disease in which the seams between the skull bones close earlier than normal, affecting the shape of the head and face.
Inherited as an autosomal dominant trait, but some cases may occur without a known family history.
Apert syndrome is caused by one of two changes to the FGFR2 gene.
The FGFR2 gene defect causes some of the bony sutures of the skull to close too early, a condition called craniosynostosis.
Early closure of sutures between bones of the skull is noted by ridging along sutures.
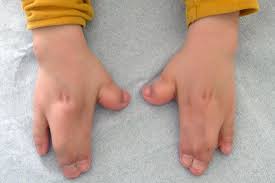
Associated with frequent ear infections, fusion or severe webbing of the 2nd, 3rd, and 4th fingers, often called “mitten hands”, hearing loss, and large or late-closing soft spot on a baby’s skull, bulging eyes, possible impaired intellectual development, under-development of the mid-face, limb skeletal abnormalities, short height, webbing or fusion of the toes.
Other syndromes can lead to a similar appearance of the face and head, but do not include the severe hand and foot features of Apert syndrome.
These similar syndromes include:
Carpenter syndrome (kleeblattschadel, cloverleaf skull deformity)
Crouzon disease (craniofacial dysostosis)
Pfeiffer syndrome
Saethre-Chotzen syndrome
Evaluation includes a physical exam, hand, foot, and skull x-rays, hearing tests and genetic testing to confirm the diagnosis of Apert syndrome.
Treatment consists of surgery to correct abnormal bone growth of the skull, as well as to repair fusion of the fingers and toes.