 Anti-synthetase syndrome is an autoimmune disease associated with interstitial lung disease, dermatomyositis, and polymyositis.
Anti-synthetase syndrome is an autoimmune disease associated with interstitial lung disease, dermatomyositis, and polymyositis.
Antisynthetase syndrome (ASS) is a multisystematic autoimmune disease associated with inflammatory myositis, interstitial lung disease, and antibodies directed against various synthetases of aminoacyl-transfer RNA.
Diagnostic criteria require one or more antisynthetase antibodies, which target tRNA synthetase enzymes, and one or more of the following three clinical features: interstitial lung disease, inflammatory myopathy, and inflammatory polyarthritis affecting small joints symmetrically.
The classic triad triad of anti-synthetase syndrome, consists of interstitial lung disease, myositis, and non-erosive arthritis, present in up to 90% of patients with AS.
Approximarely 25% of patients present only with symptoms of arthritis, 25% present with myositid and 15 to 30% have interstitial lung disease, as the presenting manifestation.
Additional conditions associated with AS include: Raynaud phenomenon, gastroesophageal reflux disease, fever, and fissured erruption of the hands.
Characterized by the presence of autoantibodies, targeting one of the aminoacyl transfer RNA synthetases.
A sub group of idiopathic, inflammatory myopathic disorders.
Autoantibodies targeting histidyltransfer RNA synthetase is also known as anti—JO-1 autoantibodies, which are the most common, with frequencies between 20 and 30% in patients with idiopathic inflammatory myopathies.
Of the 10 aminoacyl-tRNA synthetases, the most common or anti-JO-1, antibody (60%), anti-PL- 12 (17%), and anti-PL-7 (12%).
Autoantibodies are present to one of the aminoacyl-transfer RNA synthetases, which anre intracellular enzymes that anttach amino acids to RNA during protein synthesis.
AS it is associated with HLA haplotype DRB1 o301 with exposures to tobacco, cleaning chemicals, mold, bird droppings, and airborne particles.
The combination of anti-synthetase syndromes of myositis, arthritis, and interstitial lung disease is present in up to 20% of patients at disease onset, and in patients with anti-JO-1 autoantibodies can develop interstitial lung disease in up to 90% of cases.
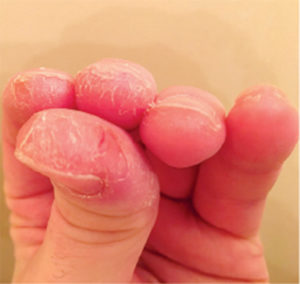
Additional features may include fever, Raynaud’s phenomenon and “mechanics hands”-thick, cracked skin usually on the palms and radial surfaces of the digits.
Antisynthetase syndrome is more prevalent in women than in men.
Interstitial lung disease may be the only manifestation of the disease. Interstitial lung disease is a major predictor of increased mortality in patients with antisynthetase syndrome.
Severe disease may develop over time.
There may be with intermittent relapses.
It is suspected that autoantibodies are formed against aminoacyl-tRNA synthetases.
Synthetases may be involved in recruiting antigen-presenting and inflammatory cells to the site of muscle or lung injury.
The most common antibody is “Anti-Jo-1” named after John P, a patient with polymyositis and interstitial lung disease/
This anti-histidyl tRNA Synthetase antibody is commonly seen in patients with pulmonary manifestations of the syndrome.
Other possible antibodies that may be seen in association with antisynthetase syndrome: Anti-PL-7, Anti-PL-12, Anti-EJ, Anti-OJ, Anti-KS, Anti-Zo, Anti-Ha-YRS, and Anti-SRP.
Evaluation:
Muscle enzymes are often elevated
Elevated levels of creatine kinase, aldolase, and inflammatory markers are seen.
Anti-Jo-1 antibody testing
Electromyography
Muscle biopsy
Pulmonary function testing
Lung biopsy
Imaging such as high resolution computed tomography.
Treatment:
Teatment for the anti-synthetase syndrome is limited, and usually involves immunosuppressive drugs such as glucocorticoids.
Additional treatment with azathioprine, methotrexate, aziothioprine, mycophenolate mofetil, cyclosporine, or tacrolimus, and nintedanib may be required in advanced cases.
Second line therapies include cyclophosphamide and biological agents such as rituximab.
CAR-T cell therapy with CD 19 targeted therapy may be effective treatment decreasing symptoms and normalization of anti-Jo-1 antibodies and pro-inflammatory cytokines interferon, Interleukin-1, IL-6 and I L-13.
Prognosis
Prognosis is largely determined by the extent of pulmonary damage.
Interstitial lung disease (ILD) affects approximately 70 to 95% of patients with a S, and is the most common cause of mobility and mortality.
CT findings include groundglass opacities, basilar fibrosis, and traction bronchiectasis.
PFTs typically show restrictive ventilation defect.
Approximately 9% of patients with zAs have rapidly progressive ILD, others have gradual onset of respiratory symptoms.
Overall survival for patients with anti-JO-1 anybody is 70% at 10 years, versus 47% with non-anti-Jo-1 synthetase antibodies.
Diagnosis requires identification of anti-synthetase auto antibodies.
If anti-synthetase syndrome is considered, a patient, should undergo high resolution CT chest and an echocardiogram to evaluate for pulmonary hypertension.
Within the first year of treatment, approximately 2/3 of patients with AS have clinical and radiologic improvement and as many as 24% a full resolution of ILD.
Approximately 1/3 of patients develop fibrotic lung disease associated with increased morbidity and mortality.
Lung transplant is considered in patients with severe or rapid progressive ILD.