 Abetalipoproteinemia is a microsomal triglyceride transfer protein (MTTP) deficiency disease.
Abetalipoproteinemia is a microsomal triglyceride transfer protein (MTTP) deficiency disease.
It is an extremely rare disorder characterized by abnormal absorption of fat and fat-soluble vitamins from food.
It is caused by a mutation in microsomal triglyceride transfer protein resulting in deficiencies in the apolipoproteins B-48 and B-100, which are used in the synthesis and exportation of chylomicrons and VLDL respectively.
It is a rare autosomal recessive disorder.
Symptoms will arise that indicate the body is not absorbing or making the lipoproteins that it needs.
Symptoms:
Failure to thrive, failure to grow in infancy.
Steatorrhea
Frothy stools
Foul smelling stools
Protruding abdomen
Intellectual disability/developmental delay
Developmental coordination disorder, evident by age ten
Ataxia
Muscle weakness
Slurred speech (dysarthria)
Scoliosis
Progressive decreased vision
Balance and coordination problems
Findings:
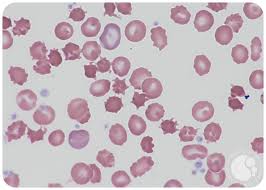
Acanthocytosis
Retinitis pigmentosa
Low blood cholesterol
It affects the absorption of dietary fats, cholesterol, and certain vitamins.
Patients affected are not able to make certain lipoproteins, which are molecules that consist of proteins combined with cholesterol and particularly triglycerides.
This leads to a multiple vitamin deficiency, affecting the fat-soluble vitamin A, vitamin D, vitamin E, and vitamin K.
Many of the observed effects are due to vitamin E deficiency, in particular.
80–99% of individuals exhibit malabsorption of fats and fat-soluble vitamins.
Approximately 30–79% of people with the disease display symptoms related to abnormality of the retinal pigmentation, ataxia, muscular hypotonia or reduced tendon reflexes.
The signs and symptoms of abetalipoproteinemia appear in the first few months of life, and include failure to gain weight and grow at the expected rate with failure to thrive, diarrhea; abnormal spiny red blood cells known as acanthocytosis, and fatty, foul-smelling stools.
The stool may contain chunks of fat and/or blood, and infants often present with gastrointestinal problems caused by the poor fat absorption, which also contributes to steatorrhea.
Other features develop later in childhood and often impair the function of the nervous system with poor muscle coordination, difficulty with balance and movement, progressive degeneration of the retina that can progress to near-blindness due to deficiency of vitamin A, retinol.
Adults may have increasing difficulty with balance and walking.
Many of the signs and symptoms of abetalipoproteinemia are a result of a severe vitamin deficiency, especially vitamin E deficiency.
Vitamin E deficiency typically results in eye problems with degeneration of the spinocerebellar and dorsal column tracts.
The MTTP gene directs the making a protein called microsomal triglyceride transfer protein, which is essential for creating beta-lipoproteins.
Beta-lipoproteins are necessary for the absorption of fats, cholesterol, and fat-soluble vitamins from the diet and necessary for the efficient transport of these substances in the bloodstream.
Most MTTP mutations of this gene lead to the production of an abnormally short microsomal triglyceride transfer protein, which prevents the normal creation of beta-lipoproteins in the body.
MTTP-associated mutations are inherited in an autosomal recessive pattern.
Abetalipoproteinemia affects multiple physiological systems.
The two most common being the nervous and the skeletal systems.
Impairment of nervous system function includes; loss of reflexes, speech impairments, tremors or involuntary motor tics, or peripheral neuropathy.
Skeletal system abnormalities often include lordosis, kyphoscoliosis, or pes cavus.
Additional complications of the diseases if not properly treated include abnormal bleeding, blindness, mental deterioration, ataxia, loss of peripheral nerve function.
Diagnosis:
Workup of Abetalipoproteinemia consists of stool sampling, a blood smear analysis and a fasting lipid panel.
Genetics test may be necessary for diagnosis.
Prenatal testing is available.
Acanthocytes are seen on blood smear.
There is no or little assimilation of chylomicrons, so their levels in plasma is low.
The inability to absorb fat in the ileum results in steatorrhea, with foul smelling stool.
There is an absence of apolipoprotein B in the blood.
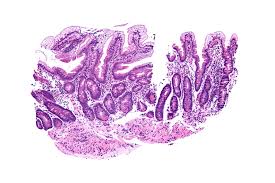
On intestinal biopsy enterocytes have vacuoles containing lipids.
Fat accumulation in the liver may occur.
Epithelial cells of the bowel lack the ability to place fats into chylomicrons, o lipids accumulate at the surface of the cell, impairing functions that are necessary for proper absorption.
Treatments:
Treatment normally consists of rigorous dieting, involving massive amounts of vitamin E.
High-dose Vitamin E therapy helps the body restore and produce lipoproteins that people with abetalipoproteinemia usually lack,and helps keep skin and eyes healthy.
Many affected males will have vision problems later on in life.
Common additional supplementation includes medium chain fatty acids and linoleic acid.
Developmental coordination disorder and muscle weakness are usually treated with physiotherapy or occupational therapy.
Dietary restriction of triglycerides is useful.
Prognosis is based on the severity of the neurological dysfunction.
Early initiation of therapy neurologic sequelae may reverse neurologic sequelae and prevent further deterioration.
Long-term outlook is reasonably good for most people when diagnosed and treated early.
Vitamin deficiencies can further compromise health.
A prolonged vitamin E deficiency can lead to the development of limiting ataxia and gait disturbances.
Some individuals may develop retinal degeneration and blindness.
If left untreated, the condition may lead to death.
A primary goal of abetalipoproteinemia is to supply fat-soluble vitamins the body lacks in the disease.