Hereditary xerocytosis (HX), also known as dehydrated hereditary stomatocytosis (DHS), is a rare genetic blood disorder where red blood cells (RBCs) become dehydrated, stiff, and prone to premature destruction.
Hereditary xerocytosis (HX) is a rare autosomal dominant hemolytic anemia caused by increased red blood cell (RBC) membrane permeability leading to cellular dehydration.
It is primarily caused by gain-of-function mutations in the PIEZO1 gene, accounting for approximately 83% of cases, or the KCNN4 gene.
Normal RBCs maintain ion balance via membrane channels.
In HX, gain-of-function mutations in PIEZO1 (mechanosensitive cation channel) — or rarely KCNN4 (Gardos channel) — cause excessive cation leak:
K⁺ leaks out faster than Na⁺ enters Net loss of intracellular cations → water follows osmotically
Result: dehydrated, dense, stiff RBCs (“xerocytes”)
These mutations cause a leak of cations—specifically potassium—out of the cell; water follows the ions, leaving the RBCs shrunken and dense.
HX often presents as a compensated hemolytic anemia, meaning the body produces new red blood cells fast enough that anemia may be mild or even absent, though evidence of cell breakdown persists.
Symptoms: Fatigue, jaundice and pale skin.
Physical Findings: Splenomegaly and a high risk of developing gallstones at a young age.
Iron Overload: Patients frequently develop significant iron buildup in organs like the liver, even if they have never received blood transfusions.
Perinatal Edema: Some infants may experience severe swelling or fluid buildup (hydrops fetalis) before or shortly after birth, which often resolves on its own.
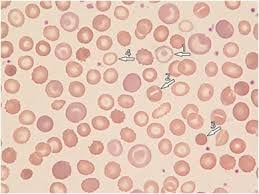
HX is frequently misdiagnosed as hereditary spherocytosis due to similar symptoms.
Blood Work often shows a high MCHC (mean corpuscular hemoglobin concentration), typically above 36 g/dL, which reflects the dense, dehydrated nature of the cells.
Osmotic gradient showing a characteristic leftward shift in cell deformability.
Sequencing of the PIEZO1 or KCNN4 genes can provide a definitive diagnosis.
Management focuses on monitoring and addressing complications rather than curing the underlying defect.
Unlike other hemolytic anemias, spleen removal is strongly avoided in HX. It does not cure the anemia and significantly increases the risk of life-threatening blood clots (thrombosis).
Folic acid supplements are often given to support RBC production.
Regular checks of ferritin levels are necessary to manage iron overload, which may require phlebotomy or chelation therapy.
Genetics Autosomal dominant inheritance PIEZO1 mutations account for ~90% of cases (chromosome 16q24.3) KCNN4 mutations cause the Gárdos channelopathy variant De novo mutations occur
Clinical Features:
Hemolysis |Mild to moderate; often compensated Anemia Typically mild; Hgb often near normal Splenomegaly Common Jaundice Episodic; worsens with infection/stress
Iron overload|Disproportionate to transfusion history Neonatal Can present with hydrops fetalis or neonatal jaundice
Laboratory Findings Elevated MCHC (>36 g/dL) — hallmark of RBC dehydration Elevated MCV with high MCHC = dense cells
Peripheral smear: target cells, stomatocytes, xerocytes
Elevated reticulocyte count
LDH, indirect bilirubin
Negative osmotic fragility (cells resist lysis — opposite of spherocytosis)
Positive osmotic gradient ektacytometry is the gold standard diagnostic test
Diagnosis Suspect ed with compensated hemolysis + high MCHC + iron overload out of proportion
Ektacytometry (osmotic gradient deformability curve shows characteristic leftward shift
Genetic testing (PIEZO1/KCNN4 sequencing)
Exclude hereditary spherocytosis (osmotic fragility is normal/decreased in HX)
Splenectomy is contraindicated or strongly discouraged in HX.
Unlike spherocytosis, splenectomy dramatically increases the risk of life-threatening thromboembolic events with portal, mesenteric, and pulmonary thrombosis.
Treatment Mostly supportive — many patients require no intervention Folic acid supplementation Monitor and treat iron overload (phlebotomy or chelation) — often develops even without transfusions due to increased GI iron absorption from ineffective erythropoiesis signals Transfusions for severe anemia/aplastic crises Avoid splenectomy
HX is likely underdiagnosed given its mild phenotype and the need for specialized testing (ektacytometry) for confirmation.